REVISIÓN BIBLIOGRÁFICA
Resistencia a la insulina como factor desencadenante de dislipidemia
Insulin resistance as a triggering factor of dyslipidemia
Roberto
Alejandro Pérez Freire1 ![]() *, Ana Gabriela Pacha Jara1
*, Ana Gabriela Pacha Jara1 ![]() *
*
1Universidad Técnica de Ambato. Facultad de Ciencias de la Salud, Carrera de Laboratorio Clínico. Ambato, Ecuador.
Citar como: Pérez Freire RA, Pacha Jara AG. Resistencia a la insulina como factor desencadenante de dislipidemia. Salud Cienc. Tecnol 2022; 2:163. https://doi.org/10.56294/saludcyt2022163
Recibido: 12-11-2022 Revisado: 26-11-2022 Aceptado: 24-12-2022 Publicado: 25-12-2022
Editor:
Prof.
Dr. Javier González Argote ![]()
RESUMEN
La resistencia a la insulina (RI) se define como un estado patológico que se caracteriza por una disminución en la sensibilidad de la insulina en los tejidos, por otra parte, la dislipidemia es una alteración a nivel lipídico que podría desarrollarse a partir de la insulinorresistencia. En esta revisión bibliográfica se describen los principales aportes científicos que demuestren si la resistencia a la insulina es una de las condiciones patológicas esenciales para desarrollar algunas de las enfermedades que constituyen graves problemas de salud pública, como la dislipidemia, diabetes mellitus tipo 2 (DM2) y el síndrome metabólico (SM).
Palabras clave: Resistencia a la Insulina; Dislipidemias; Diabetes Mellitus Tipo 2.
ABSTRACT
Insulin resistance (IR) is defined as a pathological state that is characterized by a decrease in the sensitivity of insulin in the tissues, on the other hand, dyslipidemia is a lipid level alteration that could develop from insulin resistance. This bibliographic review describes the main scientific contributions that demonstrate whether insulin resistance is one of the essential pathological conditions for developing some of the diseases that constitute serious public health problems, such as dyslipidemia, type 2 diabetes mellitus (DM2) and metabolic syndrome (MS).
Keywords: Insulin Resistance; Dyslipidemias; Type 2 Diabetes Mellitus.
INTRODUCCIÓN
La RI es una afección caracterizada por la disminución de la actividad de la insulina a nivel celular, se manifiesta en diferentes vías metabólicas, especialmente a nivel del metabolismo de la glucosa, los lípidos y las proteínas, siendo los órganos más afectados el hígado; el músculo y el tejido adiposo.(1,2)
La dislipidemia se considera como uno de los principales impulsores del desarrollo patológico de los trastornos metabólicos. Según hallazgos recientes, el tipo de dislipidemia causada por los efectos combinados de la RI y la obesidad se identificó como “dislipidemia metabólica”; definida como niveles plasmáticos marcadamente elevados de colesterol, de lipoproteínas de baja densidad (LDL-C) y triglicéridos (TG), con regulación a una disminución significativa del colesterol de lipoproteína de alta densidad (HDL-C).(3)
MÉTODOS
Se desarrolló una estrategia de búsqueda para esta revisión bibliográfica a fin de identificar los estudios apropiados, obtenidos de bases de datos electrónicas como EMBASE, PubMed y MEDLINE.
Se incluyeron aquellos artículos científicos publicados entre 2017 y 2022, mientras que se excluyeron aquellos artículos cuyo idioma no sea inglés y español, además de artículos con acceso restringido.
DESARROLLO
Insulina
La insulina es una hormona peptídica compuesta por 51 aminoácidos, producida y secretada por las células β pancreáticas, en los islotes de Langerhans. Consta de dos cadenas polipeptídicas, la cadena A contiene 21 aminoácidos y la cadena B 30 aminoácidos, que están unidas por enlaces disulfuro.(4)
La principal función de la insulina es controlar la captación, utilización y almacenamiento celular de nutrientes, aumenta el paso de glucosa sanguínea hacia los tejidos muscular y adiposo, promoviendo su conversión a glucógeno y TG, respectivamente, al tiempo que inhibe su degradación. Además, a nivel hepático, inhibe la gluconeogénesis.(4,5)
La insulina inicia su acción biológica cuando se une a los receptores específicos ubicados en la membrana celular. El receptor de insulina es una glicoproteína perteneciente a la familia de los receptores del factor de crecimiento con actividad intrínseca de Tyr quinasa (RTK's), es estimulada por sus ligandos a través de un proceso de autofosforilación en residuos de Tyr.(6,8)
El receptor insulínico es un heterotetrámero que consta de dos subunidades α y dos subunidades β, fusionadas por tres enlaces disulfuro. Las subunidades α están fuera de la membrana plasmática y poseen el sitio de unión a la insulina, y las subunidades β están divididas en tres porciones que son la extracelular, transmembrana e intracelular, sitio donde reside el dominio de actividad quinasa de Tyr.(6,8)
Cuando la insulina se une a su receptor, inicia el proceso de señalización de la insulina a través de varias interacciones proteicas. La acción de la insulina activa dos vías principales de transducción, que son la vía de las proteína cinasas activadas por mitógenos (MAP cinasas o vía MAPK), la cual regula la síntesis de proteínas; esta vía cuenta con una variada gama de posibles sustratos, incluidos los factores de transcripción que tienen participación en el proceso de regulación de la expresión génica en tejidos sensibles a la insulina, pero no en la regulación del transporte de glucosa; y la vía de señalización de la PI3K (fosfatidilinositol 3-quinasa), que es el mecanismo principal, por el cual la insulina desempeña un papel fundamental en el metabolismo de la glucosa y los lípidos, la transducción de señales a través de esta vía comienza cuando los receptores activos y autofosforilados interactúan con el sustrato del receptor de insulina (IRS) y lo fosforilan.(9,10)
Resistencia a la insulina
La RI es una condición metabólica central en la que la actividad insulínica se encuentra disminuida a nivel celular, se manifiesta en diferentes vías metabólicas, especialmente a nivel del metabolismo de la glucosa, los lípidos y las proteínas. La insulina es esencial en el metabolismo lipídico, por lo que se considera la hormona más importante que regula los procesos antilipolíticos, y los cambios de la sensibilidad celular a la insulina o los cambios de sus vías de señalización pueden afectar el metabolismo del tejido adiposo.(1,11)
En un estado de resistencia insulínica las células diana no responden a los niveles normales de insulina circulante, por lo que una respuesta normal requiere altas concentraciones de insulina. A través del tiempo se han descrito varias hipótesis para identificar los mecanismos asociados con la RI, sin embargo, la patogenia de dicha condición se puede dividir en: defectos genéticos, acumulación de lípidos ectópicos, sedentarismo, obesidad e inflamación.(13)
La actividad insulínica disminuida a nivel celular podría ser el resultado de diversas alteraciones a nivel genético, como mutaciones o modificaciones postraduccionales del receptor de insulina, el IRS.(12)
Las alteraciones más comunes en la RI son una disminución en el número de receptores de insulina y su actividad catalítica, un aumento en el estado de fosforilación de los residuos Ser/Thr de los receptores de insulina e IRS, y un aumento en la actividad de las fosfatasas del residuo Tyr, que está involucrado en la desfosforilación de los receptores y el IRS, disminución de la actividad de cinasa PI3K y Akt (enzimas que ayudan a transferir las señales en el interior de las células) y defectos en la expresión y función de GLUT-4. Estas modificaciones biologías disminuyen la incorporación de glucosa en el tejido muscular y adiposo, y favorecen las alteraciones a nivel metabólico.(12)
La inflamación es una respuesta fisiológica del sistema inmune frente a daños físicos, químicos o biológicos, se distingue por un notable aumento en el número de leucocitos, así como de citocinas proinflamatorias en la circulación o en los tejidos. La obesidad provoca cambios en los tejidos adiposo; hepático y muscular que resultan en una inflamación subclínica, que contribuye al desarrollo de RI y disfunción metabólica sistemática.(13)
Respecto a la acumulación de lípidos ectópicos, es uno de los factores de riesgo para desarrollar RI debido al aumento de ácidos grasos libres (FFA) inducidos por la obesidad, lo cual impide la señalización de insulina, por ende, la captación de glucosa en el músculo esquelético, además reduce la producción de glucógeno hepático estimulado por la insulina.(14)
Por otra parte, la señalización de la insulina aumenta el almacenamiento lipídico en los adipocitos a través de dos mecanismos: la estimulación de la síntesis de triacilglicerol, donde los TG se almacenan en pequeñas gotas de lípidos y se hidrolizan a ácidos grasos, acilglicéridos y glicerol mediante la activación de HSL (lipasa sensible a hormonas); el otro mecanismo por el que se da el aumento del almacenamiento lipídico es la inhibición de la lipólisis, que se produce al reducir los niveles de AMPc (adenosín monofosfato cíclico) e inhibir la actividad de la PKA (proteína quinasa A), en consecuencia disminuye la fosforilación de HSL, provocando la reducción en los niveles de lipólisis. La relación entre el sedentarismo y el riesgo metabólico está bien documentada, con estudios experimentales que muestran cambios nocivos en los niveles de glucosa e insulina en sangre.(13, 14,15)
Diagnóstico
En cuanto al diagnóstico, el Gold standard para la evaluación de la RI es el clamp euglucémico - hiperinsulinémico, es una técnica invasiva con más aplicación en el área investigativa que en la clínica, debido a que permite conocer la sensibilidad tisular a la insulina (tanto hepática como muscular) así como la respuesta de las células β a la glucosa.(16, 17)
Existen dos variantes de esta técnica: el clamp hiperinsulinémico, que cuantifica la utilización de glucosa en condiciones de hiperinsulinemia, y el clamp hiperglucémico, que cuantifica la respuesta pancreática de la glucosa en condiciones de hiperglucemia. Esta técnica consiste en suministrar insulina a ritmo fijo mientras se administra glucosa a un ritmo variable con el objetivo de limitar la glucemia en 90 mg/dl.(16, 17)
La aplicación de este método es compleja, laboriosa y costosa, lo que ha fomentado el desarrollo de otros métodos de evaluación de RI, como la prueba de tolerancia a la glucosa intravenosa (IVGTT) o la prueba de tolerancia a la insulina (ITT), que son usados con más frecuencia en el área diagnóstica debido a la facilidad de realizar estos test.(16, 17)
La prueba de tolerancia a la glucosa intravenosa (IVGTT) consiste en medir el nivel de insulina basal, luego administrar glucosa por vía venosa durante 3 minutos y finalmente medir los niveles de insulina en sangre 1 y 3 minutos después de la administración de glucosa; esta prueba proporciona resultados fiables ya que evita alteraciones por factores gastrointestinales en el caso de la administración de glucosa oral. La prueba de tolerancia a la insulina (ITT) mide la disminución de la glucosa en sangre después de la administración intravenosa de una determinada concentración de insulina (0,1–0,5 U/kg).(16, 17)
Los niveles de glucosa e insulina se miden cada 15 minutos, esta prueba evalúa la utilización de glucosa estimulada por la insulina por parte de los tejidos. Dado que la prueba es de corta duración, implica que hay pocas posibilidades de que las hormonas contrarreguladoras interfieran con los resultados.(16, 17)
El índice más utilizado para el diagnóstico de RI, es el modelo de homeostasis de resistencia a la insulina (HOMA-IR) debido a su simplicidad, rentabilidad y practicidad, ya que solo se requiere una muestra en ayunas. El índice HOMA-IR es un modelo propuesto por Mathews en el año 1985, se basa en la interacción entre la función de las células β pancreáticas y la sensibilidad a la insulina en un modelo matemático que utiliza las concentraciones de glucosa e insulina en ayunas.
El modelo está calibrado para una función de células β del 100 % y una RI normal de acuerdo con la siguiente fórmula:
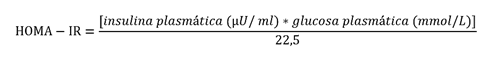
Otro de los métodos generalmente utilizados para determinar la IR es el índice QUICKI (Índice de verificación de sensibilidad a la insulina cuantitativa), basado en una ecuación logarítmica que al igual que el índice HOMA-IR también se calcula a partir de las concentraciones de glucosa e insulina en ayunas a través de la siguiente fórmula:

Debido a la amplia utilización del índice HOMA-IR como indicador de RI, varios autores han establecido un punto de corte de 2,5 en población adulta, mientras que en poblaciones pediátricas el punto de corte más utilizado es de 3,43.(18,19, 20)
Dislipidemia
La dislipidemia o hiperlipidemia es una alteración lipídica en la sangre, caracterizada por niveles elevados de colesterol (hipercolesterolemia), y una concentración elevada de TG (hipertrigliceridemia). Son condiciones frecuentes en alteraciones como; DM2, alcoholismo, insuficiencia renal crónica, hipotiroidismo, SM e insulinorresistencia. (20)
En la sangre circulan cuatro tipos principales de lípidos: colesterol, ésteres de colesterol, TG y fosfolípidos. Dada la hidrofobicidad lipídica, es necesario un medio de transporte hacia los diferentes órganos (es decir, lipoproteínas).(21,22)
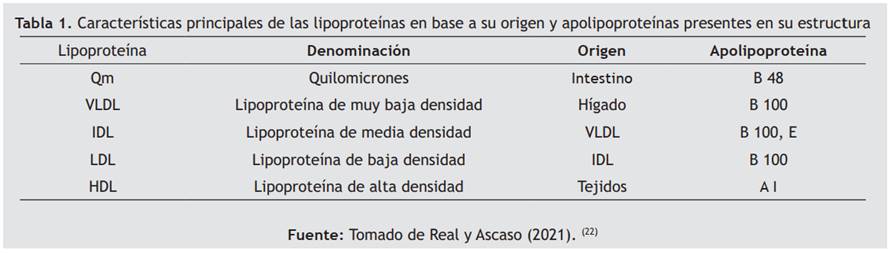
Se componen de un núcleo que contiene TG y ésteres de colesterol y una envoltura de colesterol libre, fosfolípidos y apolipoproteínas. Las lipoproteínas se clasifican en cinco clases según su densidad, origen, contenido de lípidos y contenido de apolipoproteínas. Sus respectivas características se muestran en la Tabla 1.
Las apolipoproteínas actúan como interfaces adicionales entre los lípidos y los medios acuosos y participan como activadores o inhibidores de los procesos enzimáticos del metabolismo lipídico. (21,22)
El metabolismo lipídico es un proceso desarrollado a partir de tres etapas que son:
· Transporte lipídico exógeno: transporta los lípidos de la dieta desde el intestino a varios tejidos. Los TG, colesterol y fosfolípidos del intestino se transforman en quilomicrones (QM) compuesta por apolipoproteína B 48, apolipoproteínas A-I, A-II y A-IV, que se excretan del intestino a los vasos linfáticos, alcanzando la circulación sanguínea. Aquí son hidrolizados por la lipoproteína lipasa (LPL) en el endotelio vascular, músculo y tejido adiposo, mientras que, en el hígado por la lipasa hepática (LH). A medida que circulan los QM, pierden TG, se vuelven más pequeños y densos, más ricos en colesterol y se convierten en remanentes de QM. Además, obtienen apolipoproteína C de las HDL, que es un activador de la LPL. El hígado utiliza receptores de LDL para eliminar estas partículas de la circulación. La mayoría de los TG transportados por QM se utilizan en tejidos extrahepáticos, mientras que casi todo el colesterol se devuelve al hígado.(23)
· Transporte lipídico endógeno: el hígado sintetiza colesterol a partir de ácidos grasos y TG, ambos junto con apolipoproteína B 100 que forma parte esencial de VLDL. En el plasma, pierden TG por acción de LPL y la apolipoproteína C, y se convierten en IDL. Una parte de la IDL se devuelve al hígado y otra parte se convierte en LDL. La lipoproteína de baja densidad transporta la mayor parte del colesterol plasmático y lo transfiere a los tejidos donde una parte se usa y otra se almacena como ésteres de colesterol. (23)
· Transporte de colesterol inverso: el hígado junto con el intestino sintetiza lipoproteína HDL, que atrapa el exceso de colesterol de los tejidos y otras lipoproteínas hacia el hígado durante la circulación, se metaboliza y excreta a través de los conductos biliares. (23)
Las apolipoproteínas son proteínas de la superficie lipoproteica que proporcionan estabilidad a las partículas, además de dirigir su destino metabólico, las apolipoproteínas se nombran con las letras del alfabeto y las más trascendentales son las apolipoproteínas A, B, C. La apolipoproteína A es la principal proteína responsable de mantener la estructura de las HDL y coopera con las funciones enzimáticas para transportar, reciclar y degradar el colesterol de los tejidos periféricos hasta su eliminación en el hígado.(24,25)
La apolipoproteína B es una glicoproteína anfipática que confiere estabilidad estructural a la partícula, su función es participar en la movilización del colesterol alrededor del cuerpo y es una forma de lipoproteína de baja densidad (LDL), esta apolipoproteína consta de varias isoformas, Apo B-100 compuesta por 4536 aminoácidos y sintetizada por las células hepáticas, mientras que la Apo B-48 contiene 2152 aminoácidos y es sintetizada por los enterocitos. (24,25)
La concentración de Apo B es directamente proporcional al número de partículas aterogénicas, por lo que es un marcador aterogénico, las partículas aterogénicas con Apo B son, remanentes de Qm, VLDL y sus remanentes, IDL, LDL y Lp(a).(24,25)
Por último, Apo C es una pequeña proteína de 8,8 kDa, es sintetizada por el hígado e intestino delgado y circula en asociación con VLDL, quilomicrones y HDL, esta apolipoproteína favorece la hipertrigliceridemia a través de la inhibición de la enzima lipoproteína lipasa.(24,25)
Para el diagnóstico de dislipidemia, es necesario determinar las concentraciones plasmáticas de colesterol, colesterol HDL y además de calcular el colesterol LDL, para lo cual, es recomendable utilizar la fórmula de Friedewald, dadas sus limitaciones, no se puede utilizar cuando la concentración de TG es superior a 400, y teniendo en cuenta que la concentración de colesterol LDL se puede subestimar a partir de una concentración de TG de 200 mg/dl.(26)
Una vez obtenidos los resultados del perfil lipídico básico en ayunas, se deben reconfirmar a las 2 o 3 semanas sin modificar los hábitos del paciente. También, es importante examinar la historia clínica del paciente para descartar condiciones que favorezcan al riesgo cardiovascular o que conduzcan a una posible dislipidemia primaria o congénita. Si se confirman los resultados, finalmente se puede establecer el diagnóstico de dislipidemia.(26)
Relación entre dislipidemia y resistencia a la insulina
La inhibición de la lipólisis mediada por la insulina se ve afectada cuando se desarrolla insulinorresistencia en el tejido adiposo. El aumento resultante de FFA circulantes, a su vez, empeora la RI al provocar la interrupción de la cascada de señalización de la insulina, creando un círculo vicioso.(27,28)
En el músculo, los FFA afectan la actividad PI3K asociada al sustrato del receptor de insulina (IRS-1), lo que resulta en una disminución de la translocación de GLUT-4 a la superficie, esto reduce la captación de glucosa. Al mismo tiempo, los FFA actúan a nivel hepático favoreciendo la gluconeogénesis y la lipogénesis. El resultado es un estado de hiperinsulinemia que mantiene los niveles normales de glucosa. Sin embargo, la compensación fracasa, y resulta en un descenso de los niveles de insulina, agravada aún más por el efecto lipotóxico de los FFA en las células β.(27,28)
Las elevadas concentraciones de FFA desarrollan la síntesis de ésteres de colesterol y TG, aumentando así la síntesis de lipoproteínas de muy baja densidad (VLDL) ricas en TG. Estos a su vez activan la proteína de transferencia de éster de colesterol (CETP), que favorece la transferencia de TG de VLDL a HDL, aumentando la depuración de HDL y reduciendo su concentración.(28, 29)
Además, las LDL ricas en TG formadas después del intercambio por éster de colesterol LDL, se hidrolizan mediante la lipasa hepática; lo que da como resultado una reducción en el colesterol de partículas LDL pequeñas y densas (Sd-LDL), todos estos cambios en las concentraciones de lipoproteínas son características de la dislipidemia metabólica causada por la RI.(28, 29)
El aumento de FFA conlleva a una mayor síntesis de TG y producción de apolipoproteína B, la cual está compuesta por lipoproteínas de muy baja densidad (LDL) con altas concentraciones de TG en el hígado. La elevación de Sd-LDL y la disminución del colesterol HDL son efectos indirectos de la insulinorresistencia debido al metabolismo de lípidos hepáticos alterado.(28, 29)
La dislipidemia metabólica, típica en estados de RI, obesidad, SM y DM2, implica un mayor riesgo cardiovascular, debido al estado proinflamatorio y protrombótico que también acompaña a la RI. La función endocrina del tejido adiposo encargada de la liberación de adipocinas, péptidos y citocinas inflamatorias desempeñan un papel fundamental en la fisiopatología de la RI.(28)
Los adipocitos poseen propiedades endocrinas e inmunitarias, la leptina es una adipocina liberada por el tejido adiposo visceral, su concentración se relaciona directamente con la obesidad, mayor riesgo cardiovascular e inflamación. Si las reservas de energía del cuerpo son adecuadas, la leptina inhibe la ingesta de alimentos y estimula el gasto energético, mientras controla la homeostasis de la glucosa y la sensibilidad a la insulina.(27)
Además, la leptina favorece respuestas inmunitarias proinflamatorias, ya que se ha demostrado que activa la vía Th1. Por otra parte, la adiponectina es una adipocina antiinflamatoria, antiaterogénica y antidiabética, y sus efectos contrarrestan los de la leptina. El aumento de tejido adiposo se relaciona con un déficit de adiponectina y niveles elevados de leptina, bajo este contexto incrementa el riesgo de síndrome metabólico, a su vez RI y dislipidemia.(27)
Población pediátrica
En la actualidad, existe un notable incremento de obesidad infantil a nivel mundial, alrededor de un 7 % en infantes de entre 6 y 15 años, lo cual conlleva a graves problemas de salud. La RI es importante para la obesidad; si la obesidad comienza a una edad más temprana, la edad de inicio de DM2 también es menor.
Las concentraciones séricas de colesterol total, TG y colesterol LDL, tienden a ser más altas en niños y adolescentes obesos, mientras que las concentraciones séricas de colesterol HDL son más bajas.
La RI tiene un rol importante en estas alteraciones a nivel lipídico, debido a su desarrollo en niños obesos, la lipasa sensible a hormonas y la LPL insulinodependiente no pueden inhibirse y están implicadas en la patogenia de la dislipidemia. El exceso de tejido adiposo puede provocar lipotoxicidad y liberación de citoquinas inmunitarias, ya que afecta la función y la secreción de insulina.
Un estudio realizado en 2017 evidenció que, en un grupo de niños obesos con dislipidemia, las altas concentraciones de insulina dieron como resultado un índice HOMA-IR elevado, debido a la hiperinsulinemia compensatoria que da como resultado un aumento del colesterol LDL, TG y FFA en el hígado y una disminución del colesterol HDL. Además, dicha investigación mostró que la alta incidencia de HOMA-IR fue un factor secundario importante en el desarrollo de dislipidemia, se detectaron valores más altos de HOMA-IR en niños obesos con dislipidemia y un marcado aumento en el número de casos con RI.(30)
Estudios previos también han señalado esta relación, como un estudio realizado en Holanda con 80 niños y adolescentes con obesidad severa, que demostró la presencia de factores de riesgo cardiometabólico asociados con el índice HOMA-IR.(31)
Población diabética
La DM2 es una patología metabólica, definida como el aumento de los niveles de azúcar en sangre. Suele desarrollarse por malos hábitos alimentarios y sedentarismo. La dislipidemia en DM2 presenta un grupo de alteraciones en los lípidos y lipoproteínas, conocidas como dislipidemia metabólica, caracterizada por presentar bajos niveles de colesterol de lipoproteína de alta densidad (HDL-C) y niveles elevados de lipoproteínas pequeñas de baja densidad (LDL-C).(32)
La dislipidemia, asociada a RI, es un estado frecuente que aumenta el riesgo de aterosclerosis y DM2. La dislipidemia es una alteración en el metabolismo lipídico y lipoproteico circulantes en la sangre, y en la DM2 está provocada por RI y la obesidad. La población diabética con niveles de glucosa en sangre desequilibrados tiene tendencia a desarrollar dislipidemia debido a la alta concentración de ácidos grasos libres, lo cual es un factor que implica una reducción de la sensibilidad a la insulina y el aumento de lipogénesis.(22)
Los diabéticos con niveles de glucemia descompensados son más propensos a desarrollar dislipidemia, ya que la alta concentración de FFA es un factor que implica una reducción de la sensibilidad a la insulina. La hiperglucemia favorece al desequilibrio lipídico, debido al inadecuado metabolismo lipídico.(34)
La hipertrigliceridemia es la alteración lipídica más frecuente en DM2, en una investigación realizada en el año 2014 en Brasil con una población de 339 pacientes, el 94,1 % de la muestra presentó altas concentraciones de glucosa en sangre, de los cuales el 40,7 % de los individuos no tenía dislipidemia, mientras que el 53,4 % de la muestra estudiada presentan hiperglicemia y dislipidemia asociadas.(33)
CONCLUSIONES
La resistencia a la insulina juega un papel fundamental en el desarrollo de alteraciones del metabolismo lipídico, ya que existe un incremento de lipoproteínas ricas en TG, sintetizadas por el hígado y en menor cantidad por el intestino, además de trastornos relacionados con la eliminación de estas partículas y sus remanentes.
Esta condición conlleva a un aumento en la concentración de TG, tanto en estados de ayunas como posprandiales, lo cual contribuye a una menor formación de HDL-C y a la formación de partículas de LDL pequeñas densas. También el aumento de FFA, que actúan a nivel hepático para favorecer la gluconeogénesis y la lipogénesis, conduce a una mayor síntesis de TG y producción de apolipoproteína B, compuesta por lipoproteínas de muy baja densidad (LDL) con altas concentraciones de TG en el hígado, todo este conjunto de alteraciones lipídicas termina en el desarrollo de dislipidemia metabólica.
REFERENCIAS BIBLIOGRÁFICAS
1. Lozano ES. Resistencia a Insulina: Revisión de literatura. Revista Médica Hondureña 2022;90:63-70. https://doi.org/10.5377/rmh.v90i1.13824.
2. Terlemez S, Bozdemir E, Uçar SK, Kabaroğlu C, Habif S, Kayıkçıoğlu M, et al. Insulin resistance in children with familial hyperlipidemia. Journal of Pediatric Endocrinology and Metabolism 2018;31:1349-54. https://doi.org/10.1515/jpem-2018-0337.
3. Su X, Chen X, Wang B. Pathology of metabolically-related dyslipidemia. Clinica Chimica Acta 2021;521:107-15. https://doi.org/10.1016/j.cca.2021.06.029.
4. Mujica FG. Insulina. Estructura, síntesis, secreción, depuración y degradación (Revisión). Vitae: Academia Biomédica Digital 2017:1.
5. Elías-López D, Ferreira-Hermosillo A. Insulina en poblaciones especiales: resistencia a la insulina, obesidad, embarazo, adultos mayores y enfermedad renal crónica. Revista Mexicana de Endocrinología, Metabolismo & Nutrición 2021;8:59-71. https://doi.org/10.24875/RME.M21000013.
6. Hiriart-Urdanivia M, Sánchez-Soto C, Velasco M, Sabido-Barrera J, Ortiz-Huidobro RI. El receptor soluble de insulina y el síndrome metabólico. Gac Med Mex 2019;155:541-5.
7. Posner BI. Insulin Signalling: The Inside Story. Canadian Journal of Diabetes 2017;41:108-13. https://doi.org/10.1016/j.jcjd.2016.07.002.
8. Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol 2018;19:31-44. https://doi.org/10.1038/nrm.2017.89.
9. Valero Ávila PV, Sánchez-Pérez AM. Análisis de la expresión y función de los sustratos del receptor de insulina. Agora Salud 2020;7:297-305. https://doi.org/10.6035/AgoraSalut.2020.7.30.
10. Kojta I, Chacińska M, Błachnio-Zabielska A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients 2020;12:1305. https://doi.org/10.3390/nu12051305.
11. Boucher J, Kleinridders A, Kahn CR. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb Perspect Biol 2014;6:a009191. https://doi.org/10.1101/cshperspect.a009191.
12. Gutiérrez-Rodelo C, Roura-Guiberna A, Olivares-Reyes JA. Mecanismos Moleculares de la Resistencia a la Insulina: Una Actualización. Gac Med Mex 2017;153:214-28.
13. Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuñiga FA. Association between insulin resistance and the development of cardiovascular disease. Cardiovascular Diabetology 2018;17:122. https://doi.org/10.1186/s12933-018-0762-4.
14. Carter S, Hartman Y, Holder S, Thijssen DH, Hopkins ND. Sedentary Behavior and Cardiovascular Disease Risk: Mediating Mechanisms. Exercise and Sport Sciences Reviews 2017;45:80. https://doi.org/10.1249/JES.0000000000000106.
15. Quesada MY, Hernández JC, Rode EC, Hernández OG, Bouza RC, Quesada MÁY. Índice glucosa-triglicéridos como marcador de resistencia a la insulina en pacientes con diagnóstico de hipertensión arterial esencial. Rev cubana med 2020;59:1-11.
16. Freeman AM, Pennings N. Insulin Resistance. StatPearls, Treasure Island (FL): StatPearls Publishing; 2022.
17. Placzkowska S, Pawlik-Sobecka L, Kokot I, Piwowar A. Indirect insulin resistance detection: Current clinical trends and laboratory limitations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2019;163:187-99. https://doi.org/10.5507/bp.2019.021.
18. Mirzaalian Y, Nourian M, Gholamalizadeh M, Doaei S, Hatami M, Hassanzadeh A, et al. The association of quantitative insulin sensitivity indices (HOMA-IR and QUICKI) with anthropometric and cardiometabolic indicators in adolescents. Arch Med Sci Atheroscler Dis 2019;4:e32-7. https://doi.org/10.5114/amsad.2019.84411.
19. Ruiz-Montero PJ, González-Fernández FT, Mikalacki M, Martín-Moya R. The use of anthropometrical variables for detection of homeostatic measurement assessment-insulin resistance (HOMA-IR) in female participants of a physical exercise program. Bratisl Lek Listy 2021;122:727-31. https://doi.org/10.4149/BLL_2021_116.
20. Kopin L, Lowenstein CJ. Dyslipidemia. Ann Intern Med 2017;167:ITC81-96. https://doi.org/10.7326/AITC201712050.
21. Iqbal J, Qarni AA, Hawwari A, Alghanem AF, Ahmed G. Metabolic Syndrome, Dyslipidemia and Regulation of Lipoprotein Metabolism. Current Diabetes Reviews s. f.;14:427-33. https://doi.org/10.2174/1573399813666170705161039.
22. Real JT, Ascaso JF. Metabolismo lipídico y clasificación de las hiperlipemias. Clínica e Investigación en Arteriosclerosis 2021;33:3-9. https://doi.org/10.1016/j.arteri.2020.12.008.
23. Coniglio RI. Apolipoproteína B: sus ventajas en el manejo del riesgo cardiovascular aterosclerótico. Acta bioquímica clínica latinoamericana 2021;55:11-20.
24. Carvajal Carvajal C. Los triglicéridos y la aterogénesis. Medicina Legal de Costa Rica 2017;34:82-9.
25. Candás Estébanez B, Pocoví Mieras M, Romero Román C, Vella Ramírez JC, Esteban Salán M, Castro Castro MJ, et al. Estrategia para el diagnóstico de las dislipidemias. Recomendación 2018. Laboratorio Clinico 2019;12:e21-33. https://doi.org/10.1016/j.labcli.2019.03.001.
26. Fahed G, Aoun L, Bou Zerdan M, Allam S, Bou Zerdan M, Bouferraa Y, et al. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. International Journal of Molecular Sciences 2022;23:786. https://doi.org/10.3390/ijms23020786.
27. Rochlani Y, Pothineni NV, Kovelamudi S, Mehta JL. Metabolic syndrome: pathophysiology, management, and modulation by natural compounds. Therapeutic Advances in Cardiovascular Disease 2017;11:215-25. https://doi.org/10.1177/1753944717711379.
28. Berberich AJ, Hegele RA. A Modern Approach to Dyslipidemia. Endocrine Reviews 2022;43:611-53. https://doi.org/10.1210/endrev/bnab037.
29. Ramírez JP, Leo IB, González JS, Huamán HA, Cuadros MM, Henostroza OC, et al. Obesidad, resistencia a la insulina y diabetes mellitus tipo 2 en adolescentes. Anales de la Facultad de Medicina 2018;79:200-5. https://doi.org/10.15381/anales.v79i3.15311.
30. Erol M, Bostan Gayret Ö, Hamilçıkan Ş, Can E, Yiğit ÖL. Vitamin D deficiency and insulin resistance as risk factors for dyslipidemia in obese children. Arch Argent Pediatr 2017;115:133-9. https://doi.org/10.5546/aap.2017.eng.133.
31. Castillo Núñez Y, Aguilar Salinas CA. Nutrición en diabetes con dislipidemia. Revista de la ALAD Asociación Latinoamericana de Diabetes 2017;7:31-9.
32. Feria Díaz GE, Leyva Proenza CA, Rodríguez Reyes ER, Rodríguez Moldón Y, Rodríguez Duque R, Feria Díaz GE, et al. Dislipidemia en estados de resistencia a la insulina. Correo Científico Médico 2019;23:1347-73.
33. Barbosa VS do N, Gomes LS, Palma DCA. Dislipidemia em pacientes com Diabetes tipo 2. Saúde e Pesquisa 2017;10:579-85. https://doi.org/10.17765/1983-1870.2017v10n3p579-585.
34. Cuevas M. A, Alonso K R. Dislipidemia diabética. Revista Médica Clínica Las Condes 2016;27:152-9. https://doi.org/10.1016/j.rmclc.2016.04.004.
FINANCIACIÓN
Sin financiación externa.
CONFLICTO DE INTERESES
No existen conflicto de intereses.
CONTRIBUCIÓN DE AUTORÍA
Conceptualización: Roberto Alejandro Pérez Freire, Ana Gabriela Pacha Jara.
Investigación: Roberto Alejandro Pérez Freire, Ana Gabriela Pacha Jara.
Metodología: Roberto Alejandro Pérez Freire, Ana Gabriela Pacha Jara.
Administración del proyecto: Roberto Alejandro Pérez Freire, Ana Gabriela Pacha Jara.
Supervisión: Roberto Alejandro Pérez Freire, Ana Gabriela Pacha Jara.
Redacción – borrador original: Roberto Alejandro Pérez Freire.
Redacción – revisión y edición: Ana Gabriela Pacha Jara.