REPORTE DE CASO
Síndrome de Lennox Gastaut. Reporte de caso clínico y minirevisión
Lennox Gastaut syndrome. Case report and mini-review
Marlon Andrés López García1 ![]() *, Tatiana Elizabeth Lara Abril2
*, Tatiana Elizabeth Lara Abril2 ![]() , Lissette Carolina López Lasinquiza2
, Lissette Carolina López Lasinquiza2 ![]() , Gabriela del Carmen Vargas Caiza2
, Gabriela del Carmen Vargas Caiza2 ![]() , María Belén Vargas Congo3
, María Belén Vargas Congo3 ![]()
1Solca Núcleo Tungurahua. Ambato, Ecuador.
2Hospital General Docente. Ambato, Ecuador.
3Ministerio de Salud Pública. Punibocana, Ecuador.
Citar como: López García MA, Lara Abril TE, López Lasinquiza LC, Vargas Caiza G del C, Vargas Congo MB. Síndrome de Lennox Gastaut. Reporte de caso clínico y minirevisión. Salud Cienc. Tecnol. 2022;2:170. https://doi.org/10.56294/saludcyt2022170
Recibido: 01-11-2022 Revisado: 27-11-2022 Aceptado: 20-12-2022 Publicado: 21-12-2022
Editor: Prof.
Dr. Javier González Argote ![]()
RESUMEN
Introducción: el síndrome de Lennox-Gastaut es una forma de epilepsia infantil que tiene un impacto negativo dramático en la salud física y del desarrollo del paciente. Al ser un trastorno relacionado con la edad, se caracteriza por convulsiones, electroencefalograma característico, retraso psicomotor y alteraciones del comportamiento. Es más común en los hombres y generalmente comienza antes de los ocho años y alcanza su punto máximo entre los tres y los cinco años.
Presentación del caso: se trató de un paciente de 5 años que nació prematuramente producto del cual presento secuelas de hipoxia, que alrededor de los 4 meses fue diagnosticado de epilepsia, para lo cual recibió doble terapia antiepiléptica a base de levetiracetam y ácido valproico con control parcial de sus crisis hasta antes de los 5 años, cuando presento múltiples crisis convulsivas que deterioraron la calidad de vida y lo catalogaron como síndrome de Lennox-Gastaut, en este momento fue necesario incluir un tercer fármaco a la terapia habitual llamado clobazam, para lo cual con el ajuste de dosis hasta la edad de 8 años logro presentar un mejor control sin embargo están marcadas las secuelas de parálisis cerebral infantil.
Conclusión: es uno de los síndromes más difíciles de tratar y, a menudo, es resistente a los medicamentos antiepilépticos de uso común. El pronóstico a largo plazo es malo; aunque la epilepsia suele mejorar, la ausencia total de convulsiones es rara y, en cambio, los trastornos psicológicos y psiquiátricos empeoran con el tiempo.
Palabras claves: Síndrome de Lennox-Gastaut; Encefalopatía epiléptica; Terapia antiepiléptica; Discapacidad intelectual.
ABSTRACT
Introduction: Lennox-Gastaut syndrome is a form of childhood epilepsy that has a dramatic negative impact on the physical and developmental health of the patient. Being an age-related disorder, it is characterized by seizures, characteristic electroencephalogram, psychomotor retardation and behavioral disturbances. It is more common in males and usually begins before the age of eight years and peaks between the ages of three and five years.
Case presentation: this was a 5-year-old patient who was born prematurely product of which presented sequelae of hypoxia, who around 4 months was diagnosed with epilepsy, for which he received double antiepileptic therapy based on levetiracetam and valproic acid with partial control of his seizures until before the age of 5 years, At this time it was necessary to include a third drug to the usual therapy called clobazam, for which with the adjustment of doses until the age of 8 years he achieved a better control, however the sequelae of infantile cerebral palsy are marked.
Conclusion: it is one of the most difficult syndromes to treat and is often resistant to commonly used antiepileptic drugs. The long-term prognosis is poor; although epilepsy usually improves, complete absence of seizures is rare and, instead, psychological and psychiatric disorders worsen over time.
Key words: Lennox-Gastaut Syndrome; Epileptic encephalopathy; Antiepileptic therapy; Intellectual disability.
INTRODUCCIÓN
Con una alta tasa de morbilidad y mortalidad, el síndrome de Lennox-Gastaut (SLG) es una encefalopatía epiléptica de desarrollo grave, caracterizado por múltiples tipos de convulsiones, electroencefalograma anormal (un patrón de EEG específico que muestra ráfagas de complejos de picos lentos o actividad rápida paroxística generalizada y deterioro intelectual) son sus características definitorias.(1,2,3)
La enfermedad se manifiesta por primera vez en la infancia,(4) y persisten hasta la edad adulta en más del 90 % de los pacientes.(5)
Aunque la discapacidad intelectual y los problemas de comportamiento relacionados son rasgos de este síndrome, no siempre están presentes al inicio de la enfermedad y, por lo tanto, están excluidos de los criterios de diagnóstico de la afección.(1)
Cuando se trata SLG, con frecuencia se usan en conjunto una variedad de terapias farmacéuticas y no farmacológicas. Debido a la variedad de crisis y comorbilidades con las que se asocia esta enfermedad, las decisiones terapéuticas y de manejo pueden ser un desafío. Cinco epileptólogos se reunieron para discutir un plan de tratamiento de consenso para SLG basado en los datos más recientes de una revisión de la literatura y la experiencia clínica.(1)
Se desarrollaron algoritmos de tratamiento. Como tratamiento de primera línea para pacientes con SLG de novo recién diagnosticado, las investigaciones más recientes aún recomiendan el valproato de sodio (VPA).
La lamotrigina o, posteriormente, la rufinamida se pueden usar como terapia adjunta si el VPA no es efectivo cuando se usa solo. Dada la limitada evidencia disponible, el equipo clínico debe consultar con el paciente, los padres, el cuidador y el paciente si el control de las convulsiones sigue siendo insuficiente antes de decidir sobre el próximo fármaco antiepiléptico (FAE) adicional.
El tratamiento con fármacos antiepilépticos debe considerarse junto con terapias no farmacológicas como la cirugía reconstructiva, la dieta cetogénica, la estimulación del nervio vago y la callosotomía desde el inicio del tratamiento.
La terapia con VPA debe tenerse en cuenta para los pacientes con SLG que se ha desarrollado a partir de otro tipo de epilepsia y que ya están recibiendo tratamiento con un fármaco diferente al VPA, si no lo han hecho antes. Después de eso, se debe utilizar el procedimiento para un paciente de novo.
Si es posible, solo se deben usar dos fármacos antiepilépticos simultáneamente. Se debe realizar una reevaluación exhaustiva del diagnóstico del paciente y el curso del tratamiento en pacientes con SLG establecido al menos una vez al año por un neurólogo especializado en epilepsia.(1)
CASO CLÍNICO
Se trató de un niño de 5 años nacido a las 28 semanas de gestación, por cesárea de emergencia, debido a que, presento compromiso del bienestar fetal mas gran prematuro.
En cuanto a sus antecedentes neonatales: se recepto recién nacido de sexo masculino con APGAR de 6 y 7 correspondiente al primer minuto y 5 minutos correspondiente, que necesito inmediatamente de soporte de oxigeno sin necesidad de reanimación neonatal. Además, fue necesario lavado gástrico por cantidad de meconio digerido.
Es ingresado al servicio, en donde por condición respiratoria es intubado y permaneció con esquema de antibiótico terapia durante 15 días. Posteriormente se evidencia mejoría por lo que fue retirado de ventilador mecánico y acoplan a oxigeno por Hood durante 1 mes.
Permaneció alrededor de 2 meses en el área de neonatología aparentemente egresa en condición estable, sin embargo, recién al 4 mes lactante presenta irritabilidad, desviación de la mirada hacia lado izquierdo, chupeteo, en donde acude a especialista, quien solicita electroencefalograma y tomografía axial computarizada de cráneo que revela zonas de isquemia en región temporo-parietal izquierda posteriormente diagnostica de epilepsia por encefalopatía hipóxica isquémica prescribe de levetiracetam más ácido valproico, hasta antes de los 5 años permaneció con crisis convulsivas tónico-clónicas esporádicas más parálisis cerebral infantil, siendo a esta edad en donde cuadro se exacerba con presentación de múltiples crisis mioclónicas, de ausencias, tónico clónico generalizadas, con pérdida del estado de alerta. Por ello fue necesaria la realización de nuevo electroencefalograma (Figura 1 y 2).
Figura 1. Electroencefalograma
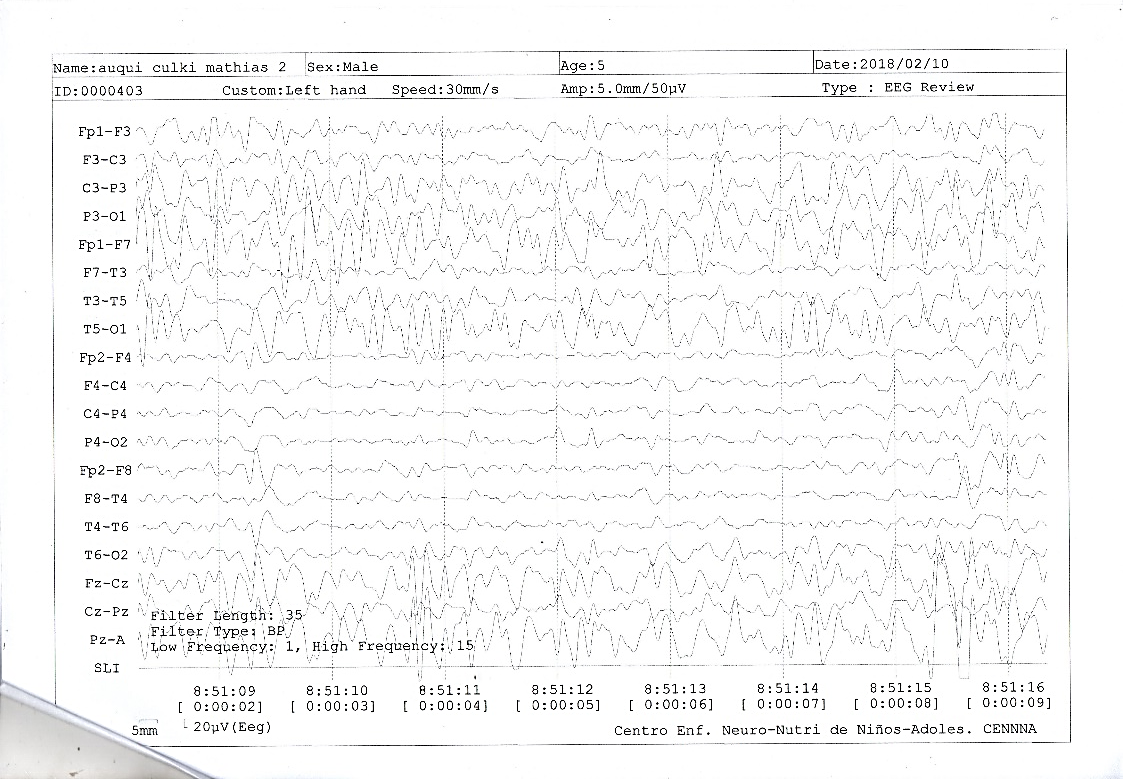
Descripción: Se observa disfunción generalizada moderada y severa actividad epiléptica generalizada de leve predominio hemisférico derecho, en patrón brote supresión, compatible con el Síndrome de Lennox-Gastaut.
Figura 2. Electroencefalograma
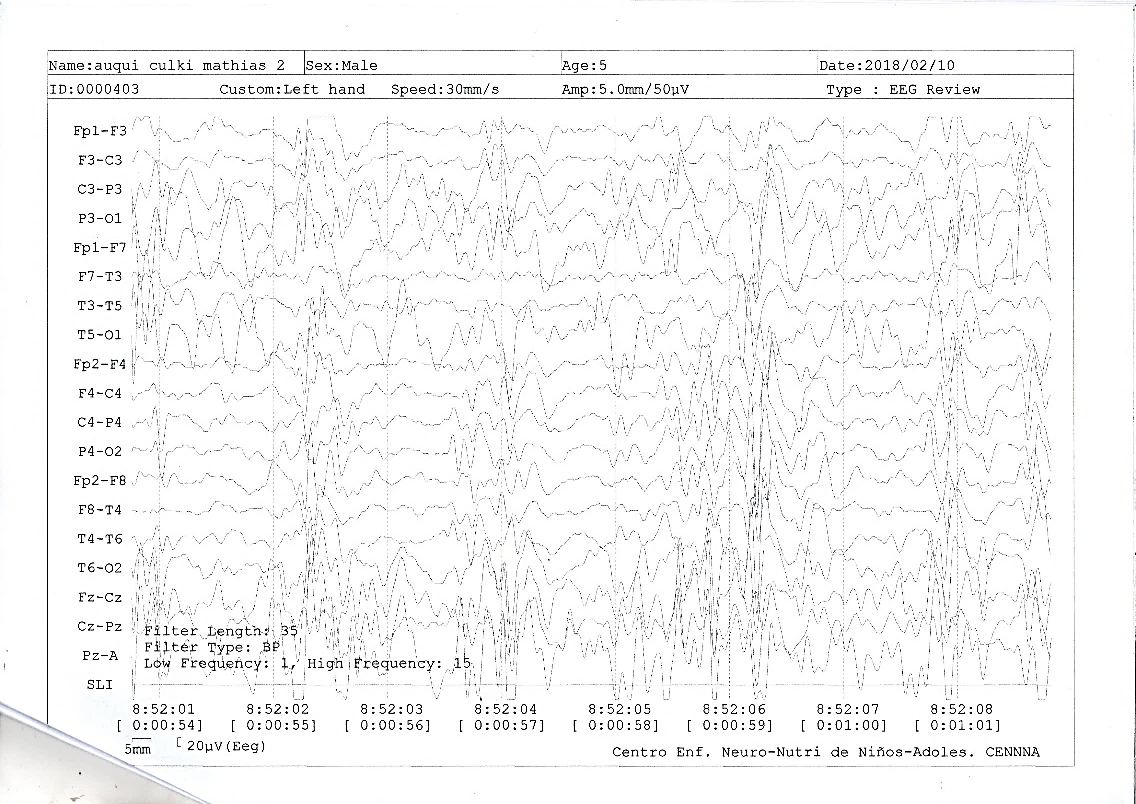
Descripción: Se observa frecuencia de base en regiones posteriores entre 6 y 6.5 Hz, correspondiente a ritmo theta, más presencia de ondas agudas de localización frontotemporal izquierdo, parietotemporal derecho y patrón brote supresión.
A los 8 años con múltiple terapia antiepiléptica compuesta por ácido valproico, clobazam, levetiracetam y para el trastorno del sueño con melatonina, presento mejoría en su electroencefalograma, caracterizando sus crisis como parciales simples (Figura 3 y 4).
Figura 3. Electroencefalograma
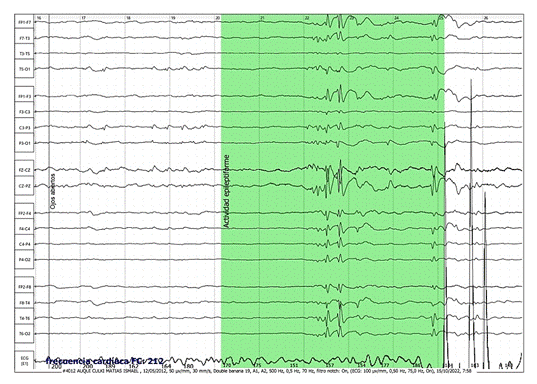
Descripción: Se observa frecuencia de base en regiones posteriores entre 5,5-6Hz, correspondiente a ritmo theta, siendo anormal para la edad.
Figura 4. Electroencefalograma
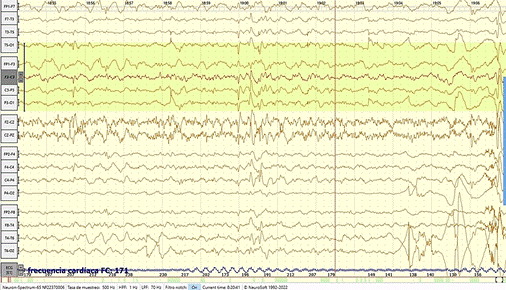
Descripción: Se observa leve actividad epileptiforme interictal focal.
DISCUSIÓN
El síndrome de Lennox-Gastaut (SLG) se considera una encefalopatía epiléptica, un concepto aprobado formalmente en 2006 por la Liga Internacional contra la Epilepsia y enfatizado aún más en 2010. El término implica que la actividad epiléptica, tanto las crisis en sí como las descargas de picos en el EEG, contribuyen al deterioro/estancamiento mental y a los trastornos de conducta.(6)
La etiología es heterogénea e incluye causas tanto genéticas como adquiridas.(7)
En cuanto a las genéticas, el número de genes identificados está creciendo rápidamente, y se ha asociado una amplia gama de mutaciones; se han descrito mutaciones y variantes en el número de copias (CNVs) que afectan al gen CHD2 (chromodomain helicase DNA-binding protein, en pacientes con encefalopatías epilépticas, discapacidad intelectual y trastornos del espectro autista, también se han visto involucrados la aparición tardía o a mutaciones patogénicas que afectan a genes como GABRB3, ALG13, SCN8A, STXBP1, DNM1, FOXG1 o CHD2. Se ha encontrado evidencia que implica a la corteza cerebral y al cuerpo calloso en la producción de algunos de los fenómenos de este síndrome.(8,9)
Los antecedentes asociados a esta patología casi siempre afectan a la corteza cerebral. Entre las causas lesionales, las que afectan a ambos lóbulos frontales son las que más comúnmente conducen al desarrollo del SLG.
Sin embargo, cualquier tipo de daño cerebral puede estar asociado y esta falta de especificidad ha impedido el esclarecimiento de los mecanismos neurofisiológicos. Algunas etiologías, como la anoxia perinatal y la encefalitis, implican múltiples niveles del sistema nervioso.(10)
El SLG es poco frecuente, con una incidencia anual de 0,2-2,8/10.000 nacimientos en los países europeos, pero su prevalencia es mayor (5 % de todas las epilepsias y alrededor del 10 % de la epilepsia infantil) debido a sus características refractarias, puede aparecer de novo en casos criptogénicos (alrededor del 30 %) o ser el resultado de lesiones cerebrales de diversas etiologías (insulto pre o perinatal, infección, diversas malformaciones incluyendo displasia y tumor cerebral) en casos sintomáticos.
En estos últimos casos, el SLG suele estar precedido (18 % - 50 %, media 30 %) por el síndrome de West o convulsiones focales. El SLG comienza entre los 2 y los 8 años (pico entre los 3 y los 5 años), ligeramente más tarde en los casos criptogénicos que en los sintomáticos, y es más frecuente en niños que en niñas.
La relación con la edad sugiere una influencia de la maduración cerebral en los síntomas y la evolución de la enfermedad. De hecho, la edad habitual de aparición se corresponde con la maduración de los lóbulos frontales y la mayoría de los signos clínicos y EEG tienen una semiología del lóbulo frontal. Recientemente, se ha sugerido un defecto en la cadena mitocondrial como causa del SLG y esta vía metabólica debería explorarse cuidadosamente en los llamados casos criptogénicos.(11)
Manifestaciones clínicas
Los múltiples tipos de crisis diarias que se producen son muy variados, suelen aparecer las crisis tónicas, que suelen ser nocturnas, las crisis atónicas (pérdidas involuntarias del tono muscular que provocan ataques de caída) o las ausencias atípicas (el niño se queda en blanco durante un minuto).
Alrededor del 60 % de los niños pueden tener crisis prolongadas o repetidas muy seguidas. Esto se denomina estado epiléptico. Algunos niños también tienen otros tipos de crisis, como mioclónicas, parciales o tónico-clónicas.(12)
Se reporta que presentan un grado de deficiencia intelectual y problemas de aprendizaje que oscila entre leve y grave, problemas de comportamiento y la depresión, que pueden atribuirse a la lesión cerebral, las frecuentes crisis, la falta de estimulación social normal o como efectos secundarios de los fármacos antiepilépticos.(12)
Como consecuencia de todas estas anomalías, el niño puede parecer irritable, cansado o aburrido. Muchos niños fracasan en la escuela y necesitan atención institucional. El desarrollo del niño rara vez es normal, y a menudo hay retraso en el desarrollo u otras formas de epilepsia. Las convulsiones pueden provocar caídas repentinas y/o pérdida de equilibrio, y se aconseja a los pacientes que lleven casco para evitar lesiones en la cabeza, la cara y los dientes.(12)
Diagnóstico
Los criterios diagnósticos clásicos del SLG consisten en una tríada de características: múltiples tipos de convulsiones, EEG anormal y deterioro cognitivo. La edad de inicio, las imágenes cerebrales anormales o normales y los factores causales no suelen considerarse importantes.
Múltiples tipos de crisis
Las crisis tónicas durante el sueño son el rasgo que suele utilizarse como base para el diagnóstico, pero, de hecho, el SLG se caracteriza por múltiples tipos de crisis concurrentes: tónicas, crisis de ausencia atípicas, atónicas y sacudidas mioclónicas.
El primer signo clínico suele ser la aparición de caídas súbitas tónicas o atónicas, denominadas "ataques de caída". Al menos el 50 % de los pacientes con SLG experimentan ataques de caída. Suelen ir precedidos de una única sacudida mioclónica generalizada seguida de una contracción tónica de los músculos axiales o atonía axial, o una combinación de ambas, que provoca una caída repentina y una lesión; en ocasiones, los pacientes llevan un casco con una máscara facial completa para evitar traumatismos craneales.
EEG anormal
Muestran complejos pico-onda lentos (originalmente conocidos como la variante petit mal) a <3 Hz que ocurren durante la vigilia (13) y es bilateralmente sincrónica, dominante sobre las regiones frontocentrales y normalmente simétrica.(14)
Los complejos consisten típicamente en un pico (duración < 70 mseg) o una onda aguda (70-200 mseg), seguida primero por un "valle" positivo profundo, y luego una onda negativa (350-400 mseg). Sin embargo, no todas las ondas están precedidas por un pico. Las ráfagas pueden aumentar y disminuir, sin un inicio y un final claros.
Deterioro cognitivo
A menudo acompañado de problemas de conducta, el deterioro cognitivo es una función de la encefalopatía epiléptica y es una característica diagnóstica esencial. Entre el 10 % y el 20 % de los niños con SLG están dentro de los rangos normales aceptados para la función cognitiva, pero tienen un procesamiento mental lento, lo que les dificulta realizar las actividades cotidianas.
La gran mayoría de los casos acaba presentando deterioro cognitivo, con una disminución del cociente intelectual (CI) a lo largo del tiempo.
Se han identificado cuatro factores de riesgo independientes para el deterioro cognitivo grave en pacientes con SLG: el estado epiléptico no convulsivo (NCSE), un diagnóstico previo de síndrome de West, una etiología sintomática de la epilepsia y una edad temprana al inicio de la epilepsia.(13)
Tratamiento
Este síndrome es una encefalopatía epiléptica crónica y compleja que se asocia con una alta tasa de resistencia a los medicamentos.
Se debe hacer un compromiso entre el control de las crisis y la prevención de los efectos adversos relacionados con la administración de fármacos antiepilépticos (FAE) para mejorar la calidad de vida de los pacientes, ya que la ausencia de crisis es un objetivo difícilmente alcanzable en el manejo clínico del síndrome.
Por ello, el tratamiento farmacológico debe personalizarse para cada paciente en función de su edad, el tipo de crisis que esté experimentando y su historial médico personal, minimizando al mismo tiempo el uso de politerapia.
· El ácido valproico (VPA) es un medicamento antiepiléptico de primera generación con un mecanismo de acción complejo y aún por dilucidar. Se cree que potencia la transmisión GABA aumentando la síntesis de GABA, reduciendo su recambio e inhibiendo su degradación, y que reduce la liberación del aminoácido excitador ácido β-hidroxibutírico; También inhibe la transmisión excitatoria mediada por el receptor N-metil-d-aspartato (NMDA), bloquea los canales de sodio activados por voltaje (VGSC) y los canales de calcio, potencia las corrientes de potasio activadas por calcio y modula la neurotransmisión serotoninérgica y dopaminérgica. El VPA ha demostrado ser útil para reducir las crisis mioclónicas, las ausencias atípicas y las crisis atónicas.
· La lamotrigina (LTG) es un medicamento antiepiléptico de segunda generación que actúa estabilizando la membrana presináptica mediante el bloqueo de los VGSC e impidiendo la liberación de neurotransmisores excitadores, principalmente glutamato.
· Rufinamida (RUF) es un medicamento antiepiléptico de tercera generación que actúa suprimiendo la excitabilidad neuronal mediante la prolongación de la inactivación de los canales de sodio activados por voltaje, lo que conduce a una disminución de la frecuencia de los disparos repetitivos sostenidos, reduce significativamente la frecuencia total de crisis, principalmente de crisis de caída. Por lo tanto, RUF sería un fármaco adecuado como segunda terapia adyuvante, una vez que VPA y LTG han fracasado.
· Topiramato (TPM) es un fármaco de segunda generación con varios mecanismos de acción; sus propiedades antiepilépticas dependen de la potenciación de la inhibición (GABA)ergica en los receptores postsinápticos GABA-A, el bloqueo de los VGSC y la supresión de los receptores AMPA/kainita. La eficacia de TPM y LTG para reducir las crisis tónicas/atónicas en pacientes con SLG.(15)
· El clobazam (CLB) es una nueva 1,5-benzodiacepina parece ejercer sus efectos aumentando los efectos inhibitorios mediados por el ácido γ-aminobutírico (GABA) a través de la unión con la subunidad α1 del receptor GABA, aumentando los transportadores de GABA e incrementando la recaptación de glutamato. El CLB se tolera bien y ha demostrado su eficacia como tratamiento complementario para diversos tipos de epilepsia, incluido el LGS.(16)
Otros tratamientos pueden incluir:(17)
· Estimulación del nervio vago;
· Dieta cetogénica;
· Benzodiacepinas como el clonazepam y el fenobarbital (fenobarbitona).
· Fármacos como la etosuximida, la metsuximida, la corticotropina (hormona adrenocorticotrópica) o los corticosteroides, la piridoxina (vitamina B6) la vigabatrina, y levetiracetam;
· Callosotomía parcial.
CONCLUSIONES
Se concluye que es un síndrome epiléptico pediátrico grave caracterizado por crisis mixtas, deterioro cognitivo y descargas generalizadas de ondas espiga lentas (<3 Hz) en la electroencefalografía.
Las crisis atónicas dan lugar a peligrosos ataques de caída con riesgo de lesiones y deterioro de la calidad de vida. Las crisis suelen ser resistentes a múltiples fármacos antiepilépticos. Actualmente se dispone de nuevos medicamentos, como la rufinamida, también en este tipo de patología se debería considerar otros tratamientos no farmacológicos como la dieta cetogénica, la estimulación del nervio vago y la cirugía de la epilepsia que incluye resección curativa focal y callosotomía.
Debido a los efectos del síndrome en el niño y su familia, se necesita un equipo de médicos especialistas para brindarle al niño el mejor control de las crisis, el más alto nivel de funcionalidad con la menor cantidad de efectos secundarios y la mejor calidad de vida posible, como, así como orientar a las familias en la dirección de los mejores recursos locales disponibles. Estos niños y sus familias requieren la asistencia de psicólogos, así como de profesionales de la salud, cuidadores, amigos y personal escolar.
REFERENCIAS BIBLIOGRAFICAS
1. Cross JH, Auvin S, Falip M, Striano P, Arzimanoglou A. Expert Opinion on the Management of Lennox–Gastaut Syndrome: Treatment Algorithms and Practical Considerations. Frontiers in Neurology 2017;8.
2. Arzimanoglou A, French J, Blume WT, Cross JH, Ernst J-P, Feucht M, et al. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. The Lancet Neurology 2009;8:82-93. https://doi.org/10.1016/S1474-4422(08)70292-8.
3. Asadi-Pooya AA. Lennox-Gastaut syndrome: a comprehensive review. Neurol Sci 2018;39:403-14. https://doi.org/10.1007/s10072-017-3188-y.
4. Brigo F, Jones K, Eltze C, Matricardi S. Anti‐seizure medications for Lennox‐Gastaut syndrome. Cochrane Database of Systematic Reviews 2021. https://doi.org/10.1002/14651858.CD003277.pub4.
5. Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, et al. Effect of Cannabidiol on Drop Seizures in the Lennox–Gastaut Syndrome. New England Journal of Medicine 2018;378:1888-97. https://doi.org/10.1056/NEJMoa1714631.
6. Archer JS, Warren AEL, Stagnitti MR, Masterton RAJ, Abbott DF, Jackson GD. Lennox-Gastaut syndrome and phenotype: Secondary network epilepsies. Epilepsia 2014;55:1245-54. https://doi.org/10.1111/epi.12682.
7. Camfield PR. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia 2011;52:3-9. https://doi.org/10.1111/j.1528-1167.2011.03177.x.
8. Lund C, Brodtkorb E, Øye A-M, Røsby O, Selmer KK. CHD2 mutations in Lennox–Gastaut syndrome. Epilepsy & Behavior 2014;33:18-21. https://doi.org/10.1016/j.yebeh.2014.02.005.
9. Mastrangelo M. Lennox–Gastaut Syndrome: A State of the Art Review. Neuropediatrics 2017;48:143-51. https://doi.org/10.1055/s-0037-1601324.
10. Blume WT. Pathogenesis of Lennox-Gastaut syndrome: Considerations and hypotheses. Epileptic Disorders 2002;3:183-96.
11. van Rijckevorsel K. Treatment of Lennox-Gastaut syndrome: overview and recent findings. Neuropsychiatr Dis Treat 2008;4:1001-19.
12. Abu Saleh T, Stephen L. Lennox gastaut syndrome, review of the literature and a case report. Head & Face Medicine 2008;4:9. https://doi.org/10.1186/1746-160X-4-9.
13. Bourgeois BFD, Douglass LM, Sankar R. Lennox-Gastaut syndrome: A consensus approach to differential diagnosis. Epilepsia 2014;55:4-9. https://doi.org/10.1111/epi.12567.
14. Markand ON. Lennox-Gastaut Syndrome (Childhood Epileptic Encephalopathy). Journal of Clinical Neurophysiology 2003;20:426.
15. Verrotti A, Striano P, Iapadre G, Zagaroli L, Bonanni P, Coppola G, et al. The pharmacological management of Lennox-Gastaut syndrome and critical literature review. Seizure 2018;63:17-25. https://doi.org/10.1016/j.seizure.2018.10.016.
16. Conry JA, Ng Y-T, Paolicchi JM, Kernitsky L, Mitchell WG, Ritter FJ, et al. Clobazam in the treatment of Lennox-Gastaut syndrome. Epilepsia 2009;50:1158-66. https://doi.org/10.1111/j.1528-1167.2008.01935.x.
17. Schmidt D, Bourgeois B. A Risk-Benefit Assessment of Therapies for Lennox-Gastaut Syndrome. Drug-Safety 2000;22:467-77. https://doi.org/10.2165/00002018-200022060-00005.
FINANCIACIÓN
Los autores no recibieron financiación para el desarrollo de la presente investigación.
CONFLICTO DE INTERESES
Los autores declaran que no existe conflicto de intereses
CONTRIBUCIÓN DE AUTORÍA
Conceptualización: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.
Curación de datos: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.
Análisis formal: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.
Investigación: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.
Administración del proyecto: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.
Recursos: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.
Supervisión: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.
Redacción –borrador original: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.
Redacción –revisión y edición: Marlon López García, Tatiana Lara Abril, Lissette López Lasinquiza, Gabriela Vargas Caiza, María Vargas Congo.