REPORTE DE CASO
Progeroid dermatological manifestations in myotonic dystrophy type 1. Case report
Manifestaciones dermatológicas progeroides en la distrofia miotónica tipo 1. Reporte de caso
Deysi Licourt Otero1
![]() *,
Melissa Toledo Licourt2
*,
Melissa Toledo Licourt2
![]() ,
Belkys Candelaria Gómez1
,
Belkys Candelaria Gómez1
![]() ,
Ilena Aurora Díaz Hernández3
,
Ilena Aurora Díaz Hernández3
![]()
1Hospital Pediátrico Provincial Docente “Pepe Portilla”. Departamento Provincial de Genética Médica. Pinar del Río, Cuba.
2Universidad de Ciencias Médicas Pinar del Río. Pinar del Río, Cuba.
3Universidad de Ciencias Médicas de Pinar del Río. Policlínico Universitario “1ero de enero”. Consolación del Sur, Pinar del Río, Cuba.
Citar como: Licourt Otero D, Toledo Licourt M, Gómez BC, Díaz Hernández IA. Progeroid dermatological manifestations in myotonic dystrophy type 1. Case report. Salud, Ciencia y Tecnología. 2024; 4:780. https://doi.org/10.56294/saludcyt2024780
Enviado: 04-12-2023 Revisado: 16-02-2024 Aceptado: 14-04-2024 Publicado: 15-04-2024
Editor: Dr.
William Castillo-González ![]()
ABSTRACT
Introduction: myotonic dystrophy type 1 is an autosomal dominant genetic disease with highly variable expressivity. Among the systemic alterations that are part of the clinical manifestations are neurodegeneration and premature aging, which is why it is part of progeroid syndromes.
Objective: to describe progeroid dermatological manifestations in type 1 myotonic dystrophy.
Case report: 44-year-old patient who, at the age of 9, confirmed a clinical diagnosis of myotonic dystrophy type 1. A family history with the same genetic disorder was collected. It presents dysmorphic signs in the skull, face, extremities and in different organs, among which are bilateral sensorineural deafness, early-onset cataracts, as well as dermatological manifestations such as seborrheic dermatitis and other lesions reminiscent of ichthyosis.
Conclusions: DM1 is often referred to as a progeroid syndrome, which implies assuming that it exposes the usual underlying mechanisms of aging that are also those that participate in the pathogenesis of DM1 and in turn justify the dermatological manifestations observed.
Keywords: Myotonic Dystrophy Type 1; Progeroid Syndrome; Seborrheic Dermatitis.
RESUMEN
Introducción: la distrofia miotónica tipo 1 es una enfermedad genética autosómica dominante con una gran expresividad variable. Entre las alteraciones sistémicas que forman parte de las manifestaciones clínicas está la neurodegeneración y el envejecimiento prematuro, es por esto que forma parte de los síndromes progeroides.
Objetivo: describir manifestaciones dermatológicas progeroides en la distrofia miotónica tipo1.
Reporte de caso: paciente de 44 años de edad que a la edad de 9 años se confirma diagnóstico clínico de distrofia miotónica tipo 1. Se recoge antecedentes familiares con igual trastorno genético. Presenta signos dismórficos en cráneo, cara, extremidades y en diferentes órganos, entre estos se destacan la sordera neurosensorial bilateral, cataratas de inicio precoz, así como las manifestaciones dermatológicas como la dermatitis seborreica y otras lesiones que recuerdan la ictiosis.
Conclusiones: la Distrofia Miotónica tipo 1, suele referirse como un síndrome progeroide, ello implica asumir que deja al descubierto los mecanismos subyacentes habituales del envejecimiento que también son los que participan en la patogénesis de la enfermedad y a su vez justifican las manifestaciones dermatológicas observadas.
Palabras clave: Distrofia Miotónica Tipo 1; Síndrome Progeroide; Dermatitis Seborreica.
INTRODUCCIÓN
En el grupo de las enfermedades neurodegenerativas y que son producidas por expansiones anormales de repetidos (conocidas como mutaciones dinámicas), se encuentra la distrofia miotónica tipo 1 (DM1) o distrofia miotónica de Steinert que se considera como la distrofia muscular más común en adultos a nivel mundial.(1) Es una enfermedad multisistémica autosómica dominante que se caracteriza principalmente por sus alteraciones neuromusculares como la miotonía y la debilidad muscular progresiva de inicio distal, defectos en la conducción cardiaca, catarata, alteraciones endocrinas, así como cierto grado de déficit cognitivo y colelitiasis.(1,2)
El gen causante de la distrofia miotónica, fue descubierto en 1992, su locus está en la región cromosómica 19q13.3. La entidad está causada por una expansión inestable del trinucleótido CTG en la región 3´ no traducida del gen DMPK (OMIM 605377), que codifica para una miosin-cinasa que se expresa principalmente en músculo esquelético.(3)
En esta enfermedad está reportada la neurodegeneración y el envejecimiento prematuro. Los procesos básicos que actúan, se consideran multifactoriales, entre los que se pueden incluir factores genéticos, ambientales y por otro lado, se encuentran los considerados endógenos que incluyen dinámicas anormales de las proteínas como degradación y agregaciones defectuosas de las proteínas, influyen además otros factores como el estrés oxidativo y formación de radicales libres funciones bioenergéticas y mitocondriales.(3,4)
La Progeria o los síndromes progeroides son condiciones que producen un envejecimiento prematuro y un acortamiento de la esperanza de vida. A diferencia del envejecimiento normal, los síndromes progeroides incluyen también signos tales como la falta de actividad ovárica o testicular (incluyendo esterilidad y ausencia de periodos menstruales) y una talla baja.(4)
La piel usualmente representa un órgano diana para expresiones genéticas severas. Algunas de estas manifestaciones clínicas en la DM1 han estado relacionadas con el envejecimiento precoz. Un contribuyente potencial al envejecimiento es la senescencia celular en la que las células del tejido envejecido experimentan una detención irreversible del crecimiento.(4,5)
El presente caso clínico tiene como objetivo, describir manifestaciones dermatológicas progeroides en el curso de la distrofia miotónica tipo 1.
REPORTE DE CASO
Se trata de un paciente masculino de 44 años de edad, piel blanca pero con múltiples lesiones. En sus primeros años de edad escolar comienza con trastornos del aprendizaje, mucho sueño durante el día y dificultades en el rendimiento intelectual por lo que no pudo terminar la secundaria básica. Refiere que lo llevaron al especialista en oftalmología porque tenía los párpados caídos y este lo remitió a Neurología quien encontró además debilidad muscular distal. Alrededor de los 9 años de edad comenzó con lesiones en piel, dadas por descamaciones, prurito y en la temporada de calor mejoraba su aspecto, pero empeoraron progresivamente. Aproximadamente a los 20 años de edad comenzó a observarse alopecia frontal asociada a otros signos dismórficos cráneo facial y esquelético y a los 32 años se diagnosticó catarata en ambos ojos, todo esto motivó su remisión al servicio de Genética Clínica. Se interrogó sobre los antecedentes familiares y se confeccionó la genealogía (figura 1) identificando varios miembros de la familia con diagnóstico clínico de Distrofia Miotónica tipo 1(DM1). A partir de este momento se convocó a varias especialidades médicas para la atención multidisciplinaria.
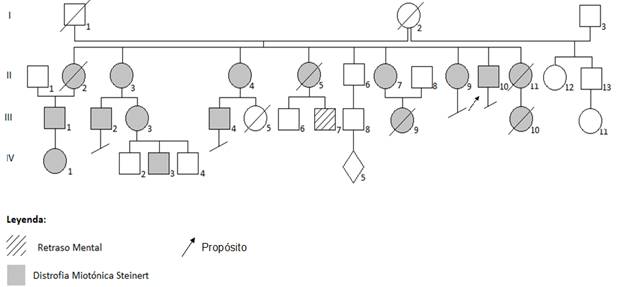
Figura 1. Árbol genealógico
Al examen físico se describen características clínicas como: cifosis, escoliosis, equinismo de miembros inferiores, rostro inexpresivo, eversión del labio inferior, labio superior en forma de v invertida, ptosis palpebral bilateral, mejillas hundidas, hipoplasia de alas nasales, columela prominente, pelo ralo grueso, alopecia frontal, dificultad para masticar, voz débil, disartria, desaparición del relieve del esternocleidomastoideo, dificultad para elevar la cabeza del plano de la cama, contracturas de codos y rodillas.
Piel: lesiones escamosas de color oscuro, secas, gruesas, grandes y generalizadas, que dan aspecto de suciedad y recuerdan la ictiosis. Presenta además múltiples pápulas y placas de superficie verrugosa en forma redondeada u ovalada, de color marrón y negro diseminadas en tronco, (Figura 2A) miembros superiores e inferiores (Figura 2B) compatibles con queratosis seborreica. Piel muy gruesa.

Figura 2. (a) Región torácica posterior con múltiples pápulas y placas de superficie verrugosa en forma redondeada u ovalada, de color marrón y negro compatibles con queratosis seborreica. (b). Miembros inferiores con lesiones escamosas, secas, gruesas, grandes que recuerda la ictiosis.
Índice de masa corporal (IMC): 17,5 bajo peso
Es un paciente que está casado pero que no ha tenido descendencia dado lo precoz de sus discapacidades. En los exámenes de laboratorio clínico solo se describen los que presenta alterados y entre paréntesis los valores de referencia.
Glicemia: 7.44 mmol/l (4,2-6,1), VLDL: 1,23 mmo/l (0,309-0,854), Triglicéridos: 1,9 mmo/l (0,68-1,88), GGT: 448,9 u/l (10-45), TGO: 116 u/l (0-49), TGP: 105,9 u/l (0-46), CPK: 498 u/l (24-195), LDH: 660 u/l (200-400), IgG: 6,1 g/l (7-16)
Informe de otros estudios realizados
EKG: Bloqueo de rama derecha y fibrilación auricular
Ecocardiograma: Disfunción diastólica
Examen oftalmológico: Cataratas bilaterales
Examen auditivo: Hipoacusia neurosensorial severa bilateral
EMG: Descargas de alta frecuencia con registro sonoro de picada de avión y prolongación del tiempo de relajación o sea el característico patrón de descarga miotónica, PUM de amplitudes disminuidas con melladuras y patrón de reclutamiento interferencial acelerado que contrasta con la debilidad al ser explorado el músculo extensor del miembro superior derecho.
Ultrasonografía abdominal: quistes renales
Resonancia magnético nuclear cerebral: múltiples lesiones hiperintensas en secuencias de TR largos (T2-FLAIR) en la sustancia blanca periventricular de ambos lóbulos temporales, frontales y periatrial bilateral algunas confluentes en forma de sierra, así como leve atrofia cerebral generalizada con dilatación exvacum secundaria del sistema ventricular.
Con todo lo anterior se realizó diagnóstico de Distrofia Miotónica tipo 1 que en su curso exhibe manifestaciones dermatológicas progeroides inusuales.
DISCUSIÓN
Las personas que viven con la DM1, constituyen una población de alto riesgo para las complicaciones multisistémicas en las que se incluye las alteraciones en la integridad de la piel.(5) En el reporte de este caso con DM1 se destaca como hallazgo inusual y precoz la presencia de diversas lesiones en la piel, unas que recuerdan la ictiosis y otras hiperpigmentadas compatibles con dermatitis seborreica así como una alopecia frontal presenil.
Hay autores que reportan una mayor prevalencia de las manifestaciones dermatológicas en pacientes con DM1. Campanati y colaboradores en estudio de casos y controles describen mayor frecuencia de la alopecia precoz (100 %) y la dermatitis seborreica (73,4 %) en pacientes con DM1 del sexo masculino y encuentran diferencias significativas entre ambos grupos para la alopecia (p<0,01), la dermatitis seborreica y las máculas hiperpigmentadas (p<0,05).(6)
Para plantear los diagnósticos dermatológicos en este paciente fue necesario descartar, mediante el método clínico, otras enfermedades de la piel en el curso de trastornos neurológicos, entre estas se encuentran las ictiosis sindrómicas como: el Síndrome de Refsum, de herencia autosómica recesiva que se manifiesta con xerosis o descamación generalizada de comienzo tardío, sin eritema, asociada a retinitis pigmentosa, neuropatía periférica, ataxia cerebelosa, anosmia, sordera progresiva y arritmias. El Síndrome KID (autosómico dominante) se caracteriza por eritroqueratodermia generalizada o localizada, hiperqueratosis puntiforme y queratodermia palmoplantar difusa. Se asocia a hipoacusia neurosensorial, queratitis, fotofobia, malformaciones cerebrales, hipotricosis, tríada de oclusión folicular, infecciones, carcinoma espinocelular. El Síndrome de Sjögren-Larsson que cursa con descamación fina generalizada, eritema e hipohidrosis asociada a paraplejía espástica, retraso mental, alteraciones oculares y su modo de herencia es autosómico recesivo.(7) Todos estos síndromes tienen en común un compromiso multisistémico en el que resaltan lo dermatológico y neurológico además de exhibir un patrón de herencia clásico en el que se pueden encontrar otros miembros de la familia afectados con iguales condiciones, no sucede esto en el paciente que se reporta en el que los antecedentes familiares son de DM1 y el paciente tiene la triada clásica característica de la enfermedad dada por miotonía, debilidad muscular y cataratas preseniles además de otros signos clínicos descritos como frecuentes, ocasionales y raros.(2,3,8)
Algunas explicaciones permiten dar respuesta a los cambios observados en la piel de este paciente, entre estas las características celulares del envejecimiento que incluyen senescencia, acortamiento de telómeros, inestabilidad genómica, disfunción mitocondrial y pérdida de proteostasis.(4,5) Las deficiencias en su control también conducen a diversos trastornos metabólicos, oncológicos, neurodegenerativos y cardiovasculares, algunos de estos están presentes en este paciente como son la hiperglicemia e hipertrigliceridemia y los trastornos de la conducción y del ritmo cardiaco (bloqueo de rama derecha y fibrilación auricular respectivamente). Se ha demostrado que la proteostasis colapsa durante el envejecimiento y hay indicios de que esto ocurre en la DM1.(8,9)
Otros estudios revelan la senescencia prematura en células de pacientes con distrofia miotónica, estos describen que las células obtenidas del músculo distal de pacientes con distrofia miotónica tipo 1 congénita, tienen una capacidad proliferativa reducida y una tasa aumentada de acortamiento de los telómeros, condicionado por mutaciones en p16. Se han reportado cambios epigenéticos dados por metilaciones en las repeticiones CTG. En el caso de la disfunción mitocondrial, se propone que los radicales libres mitocondriales causan daño oxidativo que es una fuerza impulsora del envejecimiento celular prematuro.(10,11,12)
El envejecimiento celular precoz favorece la presencia de diferentes trastornos multiorgánicos como los observados en este caso en el que resalta la expresión severa de las manifestaciones dermatológicas. Se han reportado mutaciones monogénicas que impactan, sobre varios aspectos del complejo fenotipo senescente y a las que se etiqueta como síndromes progeroides segmentarios.(4) Ellos incluyen mutaciones que resultan en inestabilidad genómica o en alteraciones del metabolismo de lípidos e hidratos de carbono. Es así como la DM1 suele referirse como un síndrome progeroide o de envejecimiento prematuro.(12)
CONCLUSIONES
Las manifestaciones clínicas multistémicas de instalación precoz y en especial la expresión severa de las alteraciones dermatológicas en el caso reportado constituyen una expresión de la DM1 que evidencia la inclusión de este trastorno entre los síndromes progeroides, ello implica asumir que deja al descubierto los mecanismos subyacentes habituales del envejecimiento que también son los que participan en la patogénesis de la DM1 y a su vez justifican las manifestaciones dermatológicas observadas.
REFERENCIAS BIBLIOGRÁFICAS
1. Ozimski LL, Sabater-Arcis M, Bargiela A, Artero R. The hallmarks of myotonic dystrophy type 1 muscle dysfunction. Biol Rev Camb Phil Soc [Internet]. 2020 [citado 20/03/2023];96(2):[aprox. 15p.]. Disponible en: https://www.academia.edu/110307761/The_hallmarks_of_myotonic_dystrophy_type_1_muscle_dysfunction?uc-sb-sw=2588012.
2. Darras BT. Myotonic dystrophy: etiology, clinical features, and diagnosis. UpToDate[Internet]. 2022 [citado 20/10/2023];22:[aprox. 6p.]. Disponible en: https://medilib.ir/uptodate/show/514
3. McKusick VA. MYOTONIC DYSTROPHY 1; DM1 [Internet]: Johns Hopkins University; 2017 [citado 20/03/2023]. Disponible en: https://www.omim.org/entry/160900.
4. Meinke P, Hintze S, Limmer S, Schoser B. Myotonic Dystrophy—A Progeroid Disease?. Front Neurol [Internet]. 2018 [citado 20/10/2023];9:[aprox. 7p.]. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6095001/.
5. Joosten I, Fuchs C, Beelen M, Plasqui G, Van Loon LJC, Faber CG. Energy Expenditure, Body Composition, and Skeletal Muscle Oxidative Capacity in Patients with Myotonic Dystrophy Type 1. J Neuromuscul Dis [Internet]. 2023 [citado 19/01/2024];10(4):[aprox. 12p.]. Disponible en: https://www.researchgate.net/publication/370570682_Energy_Expenditure_Body_Composition_and_Skeletal_Muscle_Oxidative_Capacity_in_Patients_with_Myotonic_Dystrophy_Type_1.
6. Campanati A, Giannoni M, Buratti L, Cagnetti C, Giuliodori K, Ganzetti G, et al. Skin features in myotonic dystrophy type 1: An observational study. Neuromuscular Disorders [Internet]. 2015 [citado 20/03/2023]; 25(5):[aprox. 5p.]. Disponible en: https://www.sciencedirect.com/science/article/abs/pii/S0960896615000851.
7. Vega Almendra N, Aranibar Duran L. Ictiosis hereditaria: desafío diagnóstico y terapéutico. Rev Chil Pediatr [Internet]. 2016 [citado 19/01/2024];87(3):[aprox. 11p.]. Disponible en: https://www.scielo.cl/pdf/rcp/v87n3/art13.pdf.
8. Joosten IBT, Hellebrekers DMEI, de Greef BTA, Smeets HJM, de Die-Smulders CEM. Faber CG, et al. Parental repeat length instability in myotonic dystrophy type 1 pre- and protomutations. Eur J Hum Genet [Internet]. 2020[citado 19/06/2023];28(7):[aprox. 8p.]. Disponible en: https://cris.maastrichtuniversity.nl/ws/portalfiles/portal/77657618/Faber_2020_Parental_repeat_length_instability_in.pdf.
9. Papadimas GK, Papadopoulos C, Kekou K, Kartanou Ch, Kladi A, Nitsa E, et al. A Greek National Cross-Sectional Study on Myotonic Dystrophies. Int J Mol Sci[Internet]. 2022 [citado 20/03/2023];23(24):[aprox. 7p.]. Disponible en: https://www.mdpi.com/1422-0067/23/24/15507.
10. Peric S, Pesovic J, Savic-Pavicevic D, Rakocevic Stojanovic V, Meola G. Molecular and clinical implications of variant repeats in myotonic dystrophy type 1. Int J Mol Sci [Internet]. 2021 [citado 20/03/2023];23(1):[aprox. 24p.]. Disponible en: https://www.proquest.com/openview/561957e3adfbe2a5712143b1bb4ff3cd/1.pdf?pq-origsite=gscholar&cbl=2032341.
11. Soltanzadeh P. Myotonic Dystrophies: A Genetic Overview. Genes(Basel) [Internet]. 2022 [citado 21/01/2024];13(2):[aprox. 18p.]. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8872148/pdf/genes-13-00367.pdf.
12. Wenninger S, Stahl K, Montagnese F, Schoser B. Utility and Results from a Patient-Reported Online Survey in Myotonic Dystrophies Types 1 and 2. Eur Neurol [Internet]. 2020 [citado 20/10/2023];83(5):[aprox. 10p.]. Disponible en: https://www.researchgate.net/publication/346314451_Utility_and_Results_from_a_Patient-Reported_Online_Survey_in_Myotonic_Dystrophies_Types_1_and_2.
FINANCIACIÓN
Los autores no recibieron financiación para el desarrollo de la presente investigación.
CONFLICTO DE INTERESES
Los autores declaran que no existe conflicto de intereses.
CONTRIBUCIÓN DE AUTORÍA
Conceptualización: Deysi Licourt Otero, Melissa Toledo Licourt, Belkys Candelaria Gómez, Ilena Aurora Díaz Hernández.
Investigación: Deysi Licourt Otero, Melissa Toledo Licourt, Belkys Candelaria Gómez, Ilena Aurora Díaz Hernández.
Metodología: Deysi Licourt Otero, Melissa Toledo Licourt, Belkys Candelaria Gómez, Ilena Aurora Díaz Hernández.
Administración del proyecto: Deysi Licourt Otero, Melissa Toledo Licourt, Belkys Candelaria Gómez, Ilena Aurora Díaz Hernández.
Redacción borrador original: Deysi Licourt Otero, Melissa Toledo Licourt, Belkys Candelaria Gómez, Ilena Aurora Díaz Hernández.
Redacción revisión y edición: Deysi Licourt Otero, Melissa Toledo Licourt, Belkys Candelaria Gómez, Ilena Aurora Díaz Hernández.