REPORTE DE CASO
Osteogénesis Imperfecta, a propósito de un caso
Osteogenesis Imperfecta, a case report
Gladys
Eugenia Moreno Pilozo1* ![]() ,
Andrea Verónica Castillo Ramírez2
,
Andrea Verónica Castillo Ramírez2![]() ,
Mónica Valeria Larrea Idrovo3
,
Mónica Valeria Larrea Idrovo3
![]() ,
Diana
Carolina Valle Valles4
,
Diana
Carolina Valle Valles4 ![]() ,
Luis Oswaldo Remache Guamán5
,
Luis Oswaldo Remache Guamán5![]()
1Hospital General Docente. Ambato, Ecuador.
2Hospital General Ambato IEES. Ambato, Ecuador.
3Hospital Clínica Kennedy. Ambato, Ecuador.
4Distrito de Salud 16D01 Pastaza Mera. Ambato, Ecuador.
5Hospital Divina Providencia. Ambato, Ecuador.
Citar como: Moreno Pilozo GE, Castillo Ramírez AV, Larrea Idrovo MV, Valle Valles DC, Remache Guamán LO. Osteogénesis Imperfecta. A propósito de un caso. Sal. Cienc. Tec. [Internet]. 2022 [citado Fecha de Acceso]; 2:89. Disponible en: https://doi.org/10.56294/saludcyt202289
Recibido: 07-09-2022 Aceptado: 21-10-2022 Publicado: 24-10-2022
RESUMEN
Introducción: la osteosíntesis imperfecta es una enfermedad genética rara causada principalmente por una mutación dominante en los genes que codifican para las cadenas 𝝰 del colágeno tipo I, COL1A1 y COL1A2. Dicho desorden también puede deberse a mutaciones recesivas en otros genes. Su espectro clínico incluye desde formas leves hasta formas letales perinatales. El diagnóstico clínico se puede establecer en un paciente con fracturas a repetición, escleras azules, talla baja, alteraciones de la dentición y sordera.
Reporte del caso: se trató de una paciente de 24 años tercera gesta, con antecedente obstétrico de un hijo muerto con osteogénesis imperfecta, supervisada y controlada en institución pública. A raíz de múltiples exámenes de laboratorio se corrobora, que su actual gesta presenta osteogénesis imperfecta, por lo cual se programa una cesárea a las 38,2 semanas de gestación. Durante el transoperatorio se recibe a la recién nacida viva con las características descriptas. La neonata es ingresada al servicio de neonatología donde se confirmaron las múltiples fracturas en extremidades y tórax. Ante este pronóstico reservado, la niña fallece dentro de las 24 horas por complicación pulmonar.
Conclusiones: el diagnóstico temprano en el primer trimestre del embarazo resulta indispensable para emitir un pronóstico y tratamiento oportuno en caso de ameritarlo.
Palabras claves: Osteogénesis Imperfecta; Colágeno; Huesos de cristal; Genética.
ABSTRACT
Introduction: osteogenesis imperfecta is a rare genetic disease mainly caused by a dominant mutation in COL1A1 y COL1A2 collagen type I genes which codes for collagen type I 𝝰 chains. The OI may also be the result of recessive mutations in other genes. Its clinical spectrum that goes from milder form to lethal perinatal forms. Clinical diagnosis might be established in a patient that presents repeated fractures, blue scleras, short stature, teething disorders and deafness.
Case report: the patient was 24 years old, third gestation, with an obstetric history of a dead child with osteogenesis imperfecta, who was supervised and controlled in a public institution. Several laboratory tests corroborated that the current gestation presented osteogenesis imperfecta, thus, a surgical procedure was scheduled at 38.2 weeks of gestation. During the transoperative period, we found the newborn, with the described characteristics, alive. The child was admitted to the neonatology service, where we confirmed multiple fractures in extremities and thorax. In view of this reserved prognosis, she died within 24 hours due to pulmonary complications.
Conclusions: an early diagnosis within the first three months of pregnancy is essential in order to establish a prognosis and timely treatment whenever it is necessary.
Keywords: Osteogenesis Imperfecta; Collagen; Brittle Bone Disease; Genetic.
INTRODUCCIÓN
La osteogénesis imperfecta (OI) es un grupo heterogéneo de enfermedades hereditarias, las cuales cursan con fragilidad ósea, fracturas frecuentes, deformidades óseas y talla baja.(1)
La mayoría de los pacientes muestran una mutación autosómica dominante en la cadena de colágeno tipo I alfa 1 (COL1A1) o cadena de colágeno tipo I alfa 2 (COL1A2). Sin embargo, además de los genes asociados a las cadenas de colágeno tipo I, se han identificado al menos 16 genes más los cuales estarían involucrados en el desencadenamiento de osteogénesis imperfecta. La gravedad de la presentación clínica depende del efecto de la correspondiente mutación.(2)
En lo que refiere a indicadores epidemiológicos básicos, la OI presenta una incidencia estimada de aproximadamente 1 cada 20 000 nacimientos. Respecto a su prevalencia, en Estados Unidos se estima entre los 20 000 y 50 000. Por esta razón, se la clasifica como una enfermedad huérfana.(2)
Particularmente, en Ecuador se estima que dicha condición se produce en 1 de cada 12 100-15 000 nacimientos, es decir que potencialmente habría 1 166 casos a nivel nacional. La prevalencia de OI en este país es similar no solo tanto en hombres y mujeres sino también en distintos grupos étnicos.(3)
Este conjunto de enfermedades hereditarias se presenta con una amplia variabilidad clínica que pueden incluir: baja talla, dentinogénesis imperfecta, esclera azul, pérdida auditiva, enfermedades esqueléticas, anomalías craneocervicales, asimetrías craneofaciales, desviación del tabique nasal.(4)
La insuficiencia respiratoria es una de las causas principales de mortalidad.(5) Si tras estos hallazgos existe una elevada sospecha diagnóstica, es aconsejable que el paciente sea remitido a una consulta de Genética Clínica.(1)
Si bien la manera de clasificar este desorden está en constante revisión dada su complejidad, actualmente se emplea la clasificación propuesta por Sillence en 1979. Dicha clasificación consiste en 4 tipos clínicos de OI discriminados según características clínicas, radiológicas y de severidad.(6)
En estudios realizados utilizando la secuenciación de Sanger se encontró que la Osteogénesis Imperfecta tipo I es la más frecuente, seguida de la OI tipo IV.(7)
Por otra parte, los pacientes gravemente afectados sufren múltiples fracturas espontáneas como consecuencia de traumas mínimos. En el caso de bebés con la peor forma de OI, mueren durante el período perinatal. Finalmente, las formas leves de OI pueden manifestarse solo con osteoporosis prematura o con una pérdida mineral ósea grave postmenopáusica.(8)
REPORTE DEL CASO
Se trata de una mujer de 24 años procedente y residente de Ambato, casada, instrucción secundaria, católica y de etnia mestiza; con antecedentes obstétricos G3 P0 C3 HV1 HM2 correspondientes con osteogénesis imperfecta.
Al examen físico presentó tensión arterial de 100/60 mmHg, frecuencia cardiaca de 75 latidos por minuto, frecuencia respiratoria de 17 respiraciones por minuto, temperatura de 36,5ºC, estado nutricional bueno (piel y mucosas hidratadas).
Al analizar los exámenes complementarios se determina un embarazo de 31,3 semanas de gestación, placenta fúndica posterior con madurez grado III y un peso de 1 946 gramos.
En cuanto al examen abdominal, presenta altura de fondo uterino pequeña en relación con la edad gestacional, situación dorso lateral derecho. Movimientos fetales presentes, latido cardiaco fetal de 145 latidos por minuto al tacto manual se encuentra en pelviano, no presenta contracciones uterinas. Finalmente, a nivel de la región inguinal genital no se observa secreción ni sangrado activo.
Se indicó estudio de cromosopatia, solicitado por medico particular, a las 16 semanas de gestación donde se observa un cariotipo normal para sexo femenino, riesgo de malformaciones de origen genético y multifactoriales del 4 % (figura 1). Se realizó medicación de rutina para acompañar su embarazo que consistió en suplementar con hierro y ácido fólico. Acudió al primer control a las 33,2 semanas de gestación por fecha última de menstruación (FUM) no confiable, al momento con dolor pélvico.
Figura 1. Estudio citogenético de líquido amniótico

Según una ecografía estructural se confirma el diagnóstico de osteogénesis imperfecta.
En detalle se observa: acortamiento de los fémures con deformidad y arqueamiento de los mismos e imagen en sus tercios medios que sugieren posibles fracturas, así como también acortamiento de los huesos largos de las extremidades superiores con deformidad y arqueamiento.
En el examen microscópico elemental de orina se reporta infección de vías urinarias por lo cual se instaura tratamiento antibiótico y analgésico; ecografía renal dentro de parámetros normales.
Considerando los antecedentes antes mencionados, se solicita una nueva ecografía obstétrica y se planifica la terminación del embarazo por cesárea programada a las 38.5s de gestación por FUM Vs 36.4 semanas por ecografía.
Durante el procedimiento quirúrgico se destacan los siguientes hallazgos: Líquido amniótico amarillento con mal olor, en moderada cantidad, se recibe al recién nacido de sexo femenino con Test de Apagar 5-5, peso de 2300 Gramos, talla no valorada debido a múltiples fracturas de miembros inferiores, facies sindrómicas, ausencia de huesos constituidos de cabeza (Figuras 2, 3 y 4).
Figura 2. Cavidad amniótica con tinte amarillento en moderada cantidad
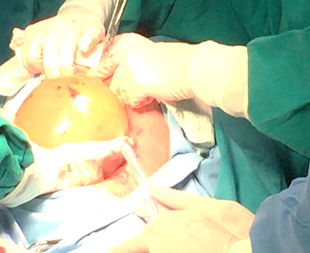
Figura 3. Extracción manual completa de saco amniótico con producto vivo
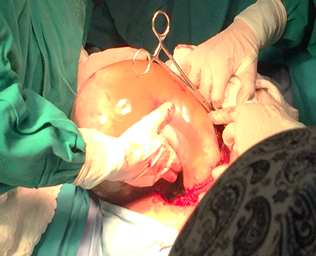
Figura 4. Recién nacida viva con osteogénesis imperfecta
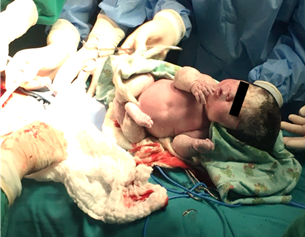
Se observó placenta fúndica posterior con presencia de un quiste cerca de cordón umbilical de aproximadamente 5 cm y anexos de características normales. La recién nacida fue trasladada al servicio de neonatología por múltiples fracturas presentadas intraútero. Falleció 24 h posterior a su nacimiento.
DISCUSIÓN
La OI es una enfermedad de herencia autosómica dominante, causada por mutaciones en los genes involucrados en la formación de colágeno. En particular, los genes COL1A1 y COL1A2 codifican para las cadenas 𝝰1 y 𝝰2 del colágeno tipo 1.(9)
Estos genes se localizan en los cromosomas 17 y 7 respectivamente. Dicha proteína es la más abundante en la piel, huesos y demás tejidos conectivos. Si bien las mutaciones en los genes indicados son responsables de más del 90 % de casos de osteogénesis imperfecta, aquellas mutaciones correspondientes a los genes CRTAP Y P3H1 se asocian con los cuadros más graves de OI.(9)
En el presente caso clínico, la sospecha diagnóstica se realizó en período prenatal valorando la ecografía obstétrica estructural donde las malformaciones esqueléticas con múltiples fracturas a nivel intrauterino determinaban un mal pronóstico. La recién nacida fallece dentro de las 24 horas por anormalidad de la arquitectura de la pared torácica que se ha descrito como una complicación grave de carácter repetitivo.(10)
Cabe destacar que se consideran hallazgos de mal pronóstico: la triada hipoplasia torácica, micromelia grave y polihidramnios siendo estas las más ominosas presentaciones de una displasia esquelética en general. En lo que refiere a la determinación de la osteogénesis imperfecta, son resulta útil realizar pruebas de imagen adicionales como: ecografías 3D, radiografías, resonancia magnética nuclear, tomografía computarizada 3D.
Otros procedimientos invasivos como la biopsia de vellosidades coriales entre las 11-14 semanas y la amniocentesis a partir de las 15-16 semanas para la obtención de ADN fetal para estudios genéticos, también pueden corroborar el diagnóstico.(11)
En ciertas ocasiones, resulta oportuno realizar paneles de genes expandidos y secuenciación del exoma o genoma con el fin de establecer el diagnóstico.(12)
En un reciente estudio de Ju y col.(13) los autores aplican el análisis de fusión de alta resolución para explorar las mutaciones COL1A1 / COL1A2, a más de confirman sus resultados mediante secuenciación de Sanger. Esta reciente técnica aumenta la precisión del diagnóstico por lo cual resulta esencial su aplicación de ser factible.
El tratamiento apunta a disminuir el riesgo de fracturas, incluir la maximización del crecimiento y la movilidad, así como manejar las complicaciones extraesqueléticas. El estándar de atención en pacientes pediátricos es la terapia con bisfosfonatos por vía intravenosa ya sea pamidronato o ácido zoledrónico antes de los 13 años.(14)
Los enfoques terapéuticos nuevos y específicos, están enfocados en inhibición de la esclerostina y la inhibición del factor de crecimiento transformante β.(12)
Pacientes tratados con ácido zoledrónico mejoraron su calidad de vida y se observa que el número de fracturas disminuyó posterior al tratamiento.(15)
CONCLUSIONES
Este caso se pudo observar como una mujer joven multípara con antecedente de un hijo muerto por osteogénesis imperfecta, que en su primera gesta no se sometió a estudios cromosómicos para establecer el posible origen del deceso, ni contaban con asesoría genética, en una tercera gesta con OI, donde no se solicitó oportunamente estudios citogenéticos ni imagenológicos.
Se evidenció como estos métodos diagnósticos nos pueden ayudar a establecer tempranamente la gravedad, pronóstico de la patología.
El estudio citogenético de líquido amniótico brindó resultados erróneos sobre bajo riesgos de malformación de origen genético y multifactorial, sin embargo, en el tercer trimestre de embarazo la ecografía estructural confirma OI y recién se pudo establecer un pronóstico y gravedad, por lo que se evaluó estrictamente hasta el final del embarazo y se planifica por antecedente quirúrgico más presentación pelviana cesárea electiva al alcanzar un embarazo a término.
En el postoperatorio se pudo determinar que según la clasificación de Sillence nos enfrentamos a una OI tipo II considerada la forma letal perinatal.
Se recomienda a los padres recibir asesoría genética y discutir la posibilidad que sus futuros embarazos podrían presentar esta condición.
Es necesario destacar que resulta importante informar en los casos más graves la posibilidad de autopsia fetal para estudios histológicos y moleculares lo que permitiría identificar su origen y conocer si esto puede influir o no en el futuro reproductivo de la pareja.
REFERENCIAS BIBLIOGRÁFICAS
1. de Pablo Márquez B, Cueto González AM, Yela Verdú C, May Llanes ME. Imperfect osteogenesis. Med Clin (Barc). 2014;143(12):e23. https://doi.org/10.1016/j.medcli.2014.05.016
2. Ohata Y, Takeyari S, Nakano Y, Kitaoka T, Nakayama H, Bizaoui V, et al. Comprehensive genetic analyses using targeted next-generation sequencing and genotype-phenotype correlations in 53 Japanese patients with osteogenesis imperfecta. Osteoporos Int. 2019;30(11):2333-42. https://doi.org/10.1007/s00198-019-05076-6
3. Ministerio de Salud Pública del Ecuador. Diagnóstico y tratamiento del paciente con osteogénesis imperfecta. Guía de Práctica Clínica. Quito: Ministerio de Salud Pública, Dirección Nacional de Normatización - MSP; 2014. Disponible en: https://sedep.com.ec/wp-content/uploads/2021/05/Diagno%CC%81stico-y-tratamiento-del-paciente-con-osteoge%CC%81nesis-imperfecta.pdf
4. Reznikov N, Dagdeviren D, Tamimi F, Glorieux F, Rauch F, Retrouvey JM. Cone-Beam Computed Tomography of Osteogenesis Imperfecta Types III and IV: Three-Dimensional Evaluation of Craniofacial Features and Upper Airways. JBMR Plus. 2019;3(6):e10124. https://doi.org/10.1002/jbm4.10124
5. Bronheim R, Khan S, Carter E, Sandhaus RA, Raggio C. Scoliosis and Cardiopulmonary Outcomes in Osteogenesis Imperfecta Patients. Spine (Phila Pa 1976). 2019;44(15):1057-63. https://doi.org/10.1097/brs.0000000000003012
6. Herreros MB, Franco R, Ascurra M. Las Osteogénesis imperfectas: revisión del tema. Pediatría (Asunción). 2008;35(1):33-7.
7. Zhytnik L, Maasalu K, Pashenko A, Khmyzov S, Reimann E, Prans E, et al. COL1A1/2 Pathogenic Variants and Phenotype Characteristics in Ukrainian Osteogénesis Imperfecta Patients. Front Genet. 2019;10:722. https://doi.org/10.3389/fgene.2019.00722
8. Gutiérrez-Díez MP, Molina Gutiérrez MA, Prieto Tato L, Parra García JI, Bueno Sánchez AM. Osteogénesis Imperfecta: Nuevas Perspectivas. Revista Española Endocrinología Pediátrica. 2013 4(160):75-85. https://doi.org/10.3266/RevEspEndocrinolPediatr.pre2013.Mar.160
9. Scaramuzzo L, Raffaelli L, Spinelli MS, Damis G, Maccauro G, Manicone PF. Orthopaedic and dental abnormalities in osteogenesis imperfecta: a review of the literature. J Biol Regul Homeost Agents. 2011;25(3):313-21.
10. Marini JC, Forlino A, Bächinger HP, Bishop NJ, Byers PH, Paepe AD, et al. Osteogénesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. https://doi.org/10.1038/nrdp.2017.52
11. Mejias Quintero ME, Salem Salem H. Osteogénesis Imperfecta. A propósito de un caso tipo II. Revista chilena de obstetricia y ginecología. 2018;83(1):86-92. http://dx.doi.org/10.4067/s0717-75262018000100086
12. Rossi V, Lee B, Marom R. Osteogenesis imperfecta: advancements in genetics and treatment. Curr Opin Pediatr. 2019;31(6):708-15. https://doi.org/10.1097%2FMOP.0000000000000813
13. Ju M, Bai X, Zhang T, Lin Y, Yang L, Zhou H, et al. Mutation spectrum of COL1A1/COL1A2 screening by high-resolution melting analysis of Chinese patients with osteogenesis imperfecta. J Bone Miner Metab. 2020;38(2):188-97. https://doi.org/10.1007/s00774-019-01039-3
14. Robinson ME, Trejo P, Palomo T, Glorieux FH, Rauch F. Osteogenesis Imperfecta: Skeletal Outcomes After Bisphosphonate Discontinuation at Final Height. J Bone Miner Res. 2019;34(12):2198-204. https://doi.org/10.1002/jbmr.3833
15. Cuevas-Olivo R, Alejo-Fuentes LJ, Alejo-Fuentes LF, Campos-Angulo G. Treatment with bisphosphonates improves the quality of life in patients with diagnosis of osteogenesis imperfecta. Acta Ortop Mex. 2019;33(2):63-6.
Los autores no recibieron financiación para el desarrollo de la presente investigación.
CONFLICTO DE INTERESES
Los autores declaran que no existe conflicto de intereses.
Conceptualización: Gladys Eugenia Moreno Pilozo, Andrea Verónica Castillo Ramírez, Mónica Valeria Larrea Idrovo, Diana Carolina Valle Valles, Luis Oswaldo Remache Guamán.
Análisis formal: Gladys Eugenia Moreno Pilozo, Andrea Verónica Castillo Ramírez, Mónica Valeria Larrea Idrovo, Diana Carolina Valle Valles, Luis Oswaldo Remache Guamán.
Investigación: Gladys Eugenia Moreno Pilozo, Andrea Verónica Castillo Ramírez, Mónica Valeria Larrea Idrovo, Diana Carolina Valle Valles, Luis Oswaldo Remache Guamán.
Metodología: Gladys Eugenia Moreno Pilozo, Andrea Verónica Castillo Ramírez, Mónica Valeria Larrea Idrovo, Diana Carolina Valle Valles, Luis Oswaldo Remache Guamán.
Recursos: Gladys Eugenia Moreno Pilozo, Andrea Verónica Castillo Ramírez, Mónica Valeria Larrea Idrovo, Diana Carolina Valle Valles, Luis Oswaldo Remache Guamán.
Visualización: Gladys Eugenia Moreno Pilozo, Andrea Verónica Castillo Ramírez, Mónica Valeria Larrea Idrovo, Diana Carolina Valle Valles, Luis Oswaldo Remache Guamán.
Redacción – borrador original: Gladys Eugenia Moreno Pilozo, Andrea Verónica Castillo Ramírez, Mónica Valeria Larrea Idrovo, Diana Carolina Valle Valles, Luis Oswaldo Remache Guamán.
Redacción – revisión y edición: Gladys Eugenia Moreno Pilozo, Andrea Verónica Castillo Ramírez, Mónica Valeria Larrea Idrovo, Diana Carolina Valle Valles, Luis Oswaldo Remache Guamán.